Chapter 7 Hearing Impairment
Hearing problems, frequent in childhood, often go unrecognized. Physicians may not inquire about hearing or routinely screen for hearing impairment. Perhaps they rely on newborn screening, overlooking the fact that it may not have been done; or perhaps a single newborn screening study was done, but progressive hearing loss was missed. Overlooking the significant hearing loss of a young child may result in a lifetime of deleterious consequences [Furth, 1966; Rapin, 1979]. The prevalence of conductive impairment resulting from chronic middle ear effusion is high among preschoolers. Its effect on language acquisition and later academic achievement is controversial, but may not be negligible in the face of another medical or social handicap [Wallace et al., 1996; Roberts and Wallace, 1997]. Universal neonatal auditory screening places the prevalence of hearing loss at 1.1 (confidence interval [CI] 0.22–3.61) per 1000 [Mehra et al., 2009], with severe to profound loss in at least 1 per 1000 children [Krahl and O’Donoghue, 2010; Fortnum et al., 2001]. The prevalence of mild hearing impairment (threshold worse than 20 decibels, the internationally accepted upper limit of normal hearing) in childhood and adolescence is 3.1 percent based on audiometric screening studies, but only 1.9 percent by self-report [Mehra et al., 2009]. Severe hearing loss in infants or toddlers precludes language learning without highly specialized and intensive habilitation and special education, unless the child happens to be the child of deaf parents whose language is sign language. Even today, most severely hearing-impaired children learn the oral and written language of their culture inadequately. The severe informational deprivation these children suffer because of inadequate language skills prevents many from achieving their cognitive and vocational potential. Early cochlear implants, which require intensive training, especially if provided later than toddlerhood, have brightened the picture, but they are not universally available or effective.
Anatomy and Physiology of the Ear and Auditory System
Except for the pinna, the entire ear is encased in the temporal bone. The ear and cranial nerve VIII provide information about two sensory modalities, sound, and the angular acceleration of the head and its position with respect to the vertical plane. Although audition and vestibular function are distinct, the inner ear contains the sensory receptor organs for both of these systems, which share a common blood supply and fluid milieu. Therefore vascular and inflammatory diseases and trauma are likely to be unselective and to affect both systems. Some genetic and biochemical defects affect audition and vestibular function selectively, others not, but the proportion of cases of vestibular impairment associated with hearing loss will remain unknown as long as vestibular function is not tested routinely in children with impaired hearing. The clinical consequences of damage to the vestibule are discussed in Chapter 8. Many excellent reviews of the anatomy and physiology of the ear are available (e.g., Brodal, 1981; Pickles, 1997; Alberti and Ruben, 1988; Wright, 1997).
Embryology and Anatomy
The inner ear
The inner ear comprises two sensory organs encased in a common fluid-filled membranous sac: the 2½ turn coiled cochlea (ventral), and the vestibule (dorsal), made up of the three semicircular canals, utricle, and saccule (Figure 7-1). The inner ear arises from the otic vesicle, the development of which begins at the end of the first month of gestation and is essentially complete in 50 days. It is encased in the otic capsule or bony labyrinth, which is adult-size at birth, lest growth alter frequency tuning of the cochlear basilar membrane. The bony labyrinth contains two separate fluid-filled compartments. The bony outer perilymphatic compartment completely surrounds the inner membranous endolymphatic compartment. The perilymph is a high-sodium plasma ultrafiltrate which, at least in infancy, communicates with the cerebrospinal fluid (CSF) through the cochlear aqueduct (see below). The auditory and vestibular receptors and first-order afferent neurons are located within the endolymphatic compartment. The endolymph is a high-potassium fluid which is maintained at +80 mV relative to the perilymph. Maintenance of this and other cellular ionic gradients is an energy-consuming process dependent on mitochondria, in particular in the highly vascularized, metabolically active stria vascularis of the scala media. These gradients are essential for generating the electrical currents that arise from the bending of the stereocilia on the endolabyrinthine vestibular and cochlear hair cells, and for reversing the ionic fluxes that bending generates. Hearing and vestibular function are dependent on the function of multiple intercellular ionic gap and tight junctions, pumps, and transporters, and on multiple neurotransmitters’ synaptic release, transporters, and receptors. Mouse models of genetic hearing losses are enormously accelerating progress in understanding the cellular and molecular biology and physiology of hearing and hearing loss [Friedman and Griffith, 2003; Frolenkov et al., 2004; Belyantseva et al., 2003; Petersen et al., 2008; Petersen and Willems, 2006; Petersen, 2002; Dror and Avraham, 2009].
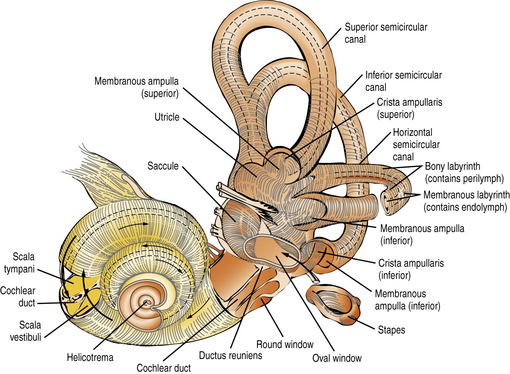
Fig. 7-1 The intricate relationship of the semicircular canals, vestibule, and cochlea.
(Courtesy of the Division of Pediatric Neurology, University of Minnesota Medical School.)
Both the perilymphatic and endolymphatic compartments have clinically significant intracranial extensions. The perilymphatic space close to the base of the scala tympani is continuous with the subarachnoid space of the posterior fossa through the cochlear aqueduct, patent in neonates but barely detected or obliterated in adults. Patency of the cochlear aqueduct in early life may be a factor in the higher prevalence of purulent endolabyrinthitis – which often results in profound deafness with vestibular impairment [Merchant and Gopen, 1996] – among young children than among adults. In children (and adults) fistulas around or through the stapes footplate may result in gushing of perilymph or even CSF into the middle ear, fluctuating, often progressive hearing loss, or episodic vertigo, and require surgical repair to prevent potential bacterial meningitis [Reilly, 1989; Weber et al., 2003]. The endolymphatic space of the vestibule, via the endolymphatic duct encased in the bony vestibular aqueduct of the otic capsule, opens into the endolymphatic sac located within the dura on the dorsal surface of the petrous bone, close to the internal acoustic meatus. Two disorders characterized by an excess of endolymphatic fluid production/pressure result in enlargement of these structures. The better-known is Ménière’s syndrome, notorious in adults but uncommon in children, which is characterized by episodes of severe vertigo and tinnitus and by a fluctuating, later progressive, sensorineural hearing loss. It may respond to diuretics, but if it does not, it may require drainage of the endolabyrinthine sac or other surgical approaches to decompress the endolabyrinthine compartment [Huang, 2002]. The second is Pendred’s syndrome, characterized by deficient transmembrane transport of chloride and iodide anions, causing a severe prelingual hearing loss and adolescent or adult euthyroid goiter. Knockout mice deficient in pendrin have a decreased endolymphatic potential, the pH is lowered, and Ca++ is increased [Nakaya et al., 2007]. Fetal enlargement of the endolabyrinthic duct and sac can be presumed to be the reason for congenital enlargement of the vestibular aqueduct (EVA) or other malformations of the otic capsule (Mondini malformation) frequent in Pendred syndrome but also seen in other syndromes [Petersen and Willems, 2006; Gonzalez-Garcia et al., 2006].
The scala media, in which the organ of Corti resides, is triangular in cross-section, with its apex directed medially toward the modiolus (axis) of the cochlea, to which it is anchored. Its vertical side, mostly lined by the stria vascularis, is apposed to the osseous outer wall of the cochlea (Figure 7-2). The floor of the scala media is formed by the basilar membrane and osseous spiral lamina, which separate the scala media from the scala tympani. The roof of the scala media is formed by Reissner’s membrane, partitioning it from the scala vestibuli. The organ of Corti sits on the basilar membrane, a coiled, mobile, truncated triangular membrane whose stiffer, narrower end is located at the base of the cochlea and whose wider, more pliable end is located at the apex. The organ of Corti consists of a long, orderly, longitudinal file of radial (transverse) rows. Each row is made up of four hair cells surrounded by specialized supporting cells located on either side of the hollow tunnel of Corti. Adjacent rows are stimulated by maxima of the traveling wave and are thus narrowly tuned to particular sound frequencies. Two pillar cells meeting at the apex of the triangular tunnel make up its lateral walls and the basilar membrane its floor; the tunnel is filled by the cortilymph, which has its own ionic composition. Each radial row comprises a single inner hair cell situated on the modiolar side of the tunnel and three efferent outer hair cells. The inner hair cell is the auditory afferent receptor, of which there are about 3500 [Spoendlin, 1988].
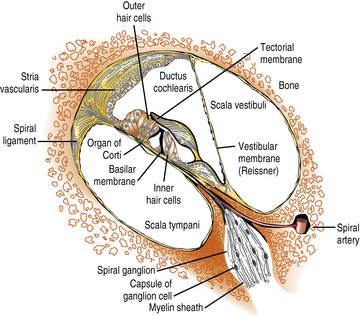
Fig. 7-2 Structural components of the organ of Corti.
(Courtesy of the Division of Pediatric Neurology, University of Minnesota Medical School.)
There are about 12,000 efferent outer hair cells, which receive their inputs from the olivocochlear bundle. Their role is to modulate acoustic reception by the inner hair cells and spiral ganglion cells [Hunter-Duvar and Harrison, 1988]. On its apical surface, each inner hair cell has a straight palissade of some 60 stereocilia, whereas the palissade is arranged in a V shape on each outer hair cell. Three stereocilia of graded length, linked at their tips, make up each “stick” of the palissades, an arrangement that ensures that only the longest cilium of the outer hair cells reaches the gelatinous tectorial membrane attached to the modiolus. Stereocilia contain actin filaments and myosins and other motor molecules [Dror and Avraham, 2009]. The hair cells and supporting cells thus tightly couple movements of the basilar and tectorial membranes, ensuring synchronized bending of the stereocilia in response to the traveling waves induced by sound along the basilar membrane [Frolenkov et al., 2004]. Cell adhesion proteins (cadherins) keep hair cells, cilia, and supporting cells tightly packed, and ionic channels provide fast communication among them. The base of each inner hair cell synapses with the dendrites of about 20 type I spiral ganglion neurons, which number about 30,000, whereas the dendrites of a single smaller type II spiral ganglion neuron contact about 10 outer hair cells. Axons of the efferent olivocochlear bundle divide widely, each synapsing on many outer hair cells; in contrast, they terminate sparingly on the inner hair cells and dendrites of the type I spiral ganglion neurons [Hunter-Duvar and Harrison, 1988]. Glutamate is the main excitatory neurotransmitter between the inner hair cells and ionotropic receptors on the dendrites of type I spiral auditory neurons [Oestreicher et al., 2002]. Olivocochlear synapses of the efferent pathway with the outer hair cells and type II spiral ganglion neurons are mainly inhibitory through the release of gamma-aminobutyric acid (GABA) and dopamine, although its modulation also involves release of acetylcholine and other excitatory neurotransmitters.
Central auditory pathway
The axons of the spiral ganglion neurons project to the cochlear nuclei of the medulla. They are of quite uniform caliber, which means that their conduction velocities are uniform, crucial for sound transmission. Axons from the base of the cochlea, which carry high-frequency sounds, project to the ventral cochlear nucleus and ventral portion of the dorsal cochlear nucleus; those from the apex of the cochlea, which carry low-frequency sounds, project to the dorsal portion of the dorsal cochlear nucleus (Figure 7-3). Each spiral ganglion neuron is connected to five different types of neurons in the cochlear nuclei. Most axons of the cochlear nuclei cross the midline, forming the acoustic striae on the floor of the fourth ventricle as they course through the trapezoid body to form the ascending lateral lemniscus. Relays in the central auditory pathway include the cochlear nuclei, superior olivary complex, nucleus of the lateral lemniscus, inferior colliculus, and medial geniculate body of the thalamus where all fibers synapse before reaching the primary and secondary auditory cortices of the superior temporal gyrus. Tremendous multiplication characterizes the auditory pathway of the rhesus monkey, which goes from the 3,500 inner hair cells to 30,000 spiral ganglion neurons and to about 10 million neurons in the medial geniculate body, which projects to both primary (area 41) and secondary (area 42) auditory cortices in the superior temporal gyrus. Neurons of area 41 project to area 42 situated behind it. The secondary auditory areas of the two hemispheres are connected through fibers traveling in the body of the corpus callosum. They are also widely connected to other cortical areas, including frontal and temporoparietal language areas, the cingulate gyrus, and frontolateral cortices.
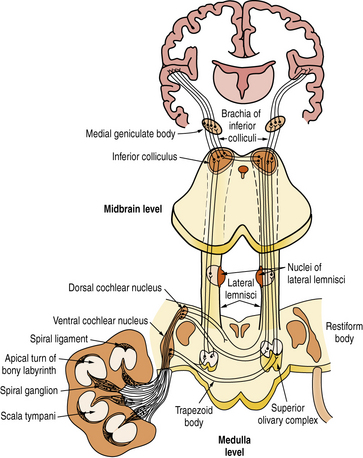
Physiology
The relevant dimensions of sound are intensity, which is expressed in decibels (dB), and frequency, which is measured in cycles per second or Hertz (Hz) [Katz, 2002]. Units of intensity and frequency are logarithmic. The decibel scale displayed on audiograms appears to be linear, but the units themselves are logarithmic. An increase of about 10 dB results approximately in doubling of subjective loudness. Audiometers are calibrated so that audiometric 0 (i.e., normal auditory sensitivity or hearing level, or 0 dB of hearing loss) corresponds to the internationally accepted average threshold of normal hearing at each frequency. The lower limit of normal hearing has been set at 20 dB below audiometric 0 across the frequency range. The ear is most sensitive at about 2000 Hz and very sensitive between 1000 and 4000 Hz, with sensitivity decreasing rapidly below 500 Hz (low frequencies/pitches) and above 4000 Hz (high frequencies/pitches). At the extremes of the audible frequency band, more sound pressure is required to reach threshold than in the middle of the band. Therefore, the sensitivity curve across frequencies of the human ear is U-shaped; for the sake of convenience, this was transformed to a straight line representing mean normal hearing (sound pressure associated with 0 dB of hearing loss). Localization of sound in space is in part mediated by differences in intensity and lag in phase of sounds reaching each of the ears. Processing of sound frequency, intensity, location, onset, and cessation takes place largely in brainstem auditory relays.
The primary role of the pinna is in sound localization. The middle ear acts as a sound amplifier that compensates for loss of sound energy as it is transmitted from air to fluid because of the much greater area of the tympanic membrane compared to that of the oval window, the vibration pattern of the eardrum, and the leverage of the ossicular chain. Sound in air is also transmitted to the cochlea through the skull, but it occurs at threshold intensities that exceed air conduction by 40–60 dB. Bone conduction is important in children with prolonged or frequent middle ear effusions, whose thresholds may be raised up to 40–60 dB at times. Middle ear effusion may be detrimental for language acquisition if associated with other social or medical handicaps [Wallace et al., 1996], but it never renders children profoundly deaf. Bone conduction prostheses are critical for children with anotia, canal atresia, and other malformations of the external and middle ear who may have normally functioning cochleas and can therefore acquire language competently if provided promptly with appropriate bone conduction hearing aids or other prostheses.
Classification of Hearing Losses
Multiple overlapping, non-mutually exclusive clinical dimensions are required to define and classify hearing losses. Each clinically derived subtype encompasses multiple etiologies (Box 7-1).
Box 7-1 Likely Causes of Chronic Hearing Loss Classified by Type of Hearing Loss





Sensorineural (Endocochlear) Hearing Loss





















Classification of Hearing Loss by Clinical Features
Sensorineural (Endocochlear) Hearing Loss
Sensorineural hearing loss may or may not be associated with vestibular dysfunction. Vestibular dysfunction was present in 41 percent of pupils at a Dutch school for the deaf, and was most prevalent in children with thresholds above 90 dB and those with exogenous hearing losses [Huyghen et al., 1993]. It is characteristic of Usher’s syndrome [Petersen and Willems, 2006; Ahmed et al., 2009] and of many children deafened by purulent meningitis [Merchant and Gopen, 1996]. It is not clear how much an awareness of the presence or absence of a vestibular deficit would help in the differential diagnosis of congenital hearing loss because vestibular testing is not performed routinely in deaf children.
Sensory hearing loss
This term refers to selective pathology of the inner hair cells, such as their inadequate development, dysfunction, degeneration, or death. Some pathologies may preclude hearing from the start, while others may be progressive; the speed of progression will determine the age at which hearing will become functionally compromised. Many of the consequences of genetic hearing loss (discussed later) are very specific. Some are selective for the cilia, which can be malformed, have impaired motility, be disorganized rather than neatly bundled, or have faulty intercommunication or connections with the tectorial membrane. Ciliated receptor cells of the labyrinth and retinal rods and cones may also be involved by the same mutations, resulting in the three subtypes of Usher’s syndrome [Friedman and Griffith, 2003; Petersen and Willems, 2006; Dror and Avraham, 2009]. Although most pathologies involve both inner and outer hair cells, a few are selective for the inner hair cells, so that OAE testing of the outer hair cells as the sole screening surrogate for inner hair function can be severely misleading, as was the case in a few premature infants whose temporal bones were examined postmortem [Amatuzzi et al., 2001]. One of many mutations of the GJB2 gene results in lack of the connexin 26 K+ gap-junction protein, and is responsible for almost half the cases of profound nonsyndromic recessive prelingual hearing loss. Depending on the amount of connexin 26 protein synthesized, however, some GBJ2 mutations may be selective for the inner hair cells, with resultant absent ABRs but misleadingly preserved OAEs, the pattern audiologists label “auditory neuropathy” (see below). Inadequate synthesis of claudin, an adhesion molecule of intercellular tight junctions, results in a rare recessive deafness due to selective degeneration of the outer hair cells. Some other hearing loss is associated with initially nonlethal malfunction of ionic pumps and channels or neurotransmitter release, or with faulty transporters, receptors, or reuptake at synapses between the hair cells and dendrites of the spiral ganglion cells. For example, there are mutations of genes for collagen which affect the tectorial membrane. In short, as more and more different mutations are being identified that involve not only the hair cells but also other cells and tissues in the organ of Corti, endocochlear hearing loss may be a more accurate term than sensory hearing loss. To complicate matters further, what starts out as a sensory (hair cell) deficit may not remain “pure”. Deafferentation of the spiral ganglion cells may eventually lead to their apoptic death, and vice versa [Sobkowicz et al., 1999]; spiral ganglion neurons receive both afferent (hair cell) and efferent (olivary, outer hair cell) innervation [Sobkowicz et al., 2004]. Trans-synaptic degeneration may not be inevitable in humans [Teufert et al., 2006], although the endstage of long-standing hearing loss is often collapse of the entire organ of Corti and depletion of neuronal cell bodies in the modiolus, including their axons in the eighth nerve, with trans-synaptic degeneration in higher auditory relays [Rapin et al., 2006; Gandolfi et al., 1984].
Hearing loss due to pathology in spiral ganglion neurons: “auditory neuropathy”
Dysfunction at the synapse between the inner hair cells and type I spiral ganglion cells may arise from problems with the inner hair cells’ release or reuptake of neurotransmitter, with transporters, or with the glutamate receptors or ion-gated channels of the myelinated dendrites of the spiral ganglion cells. For example, otoferlin deficiency causes profound deafness because of impaired vesicle exocytosis specific to these synapses [Santarelli et al., 2008]. It fulfills audiologic criteria for an audiologic diagnosis of “auditory neuropathy”, i.e., absent ABRs and preserved OAEs, about which two points need to be made. First, this test result alone does not allow a distinction to be made between malfunction of the inner hair cells, type I spiral ganglion neurons, or the synapses between them. Second, hearing loss may be missed if one relies on OAEs without ABRs because OAEs reflect only outer hair cell function. Pathology of spiral ganglion cell soma results in degeneration of both their dendrites and axons, a classic afferent axonal auditory neuropathy with an inexcitable eighth nerve. Compression of the eighth nerve, or a biochemical or autoimmune disorder of myelin affecting both dendrites and axons, will slow and dys-synchronize conduction, with a resultant demyelinating auditory neuropathy. Long-standing axonal and demyelinating neuropathies ultimately become mixed and the nerve becomes inexcitable.
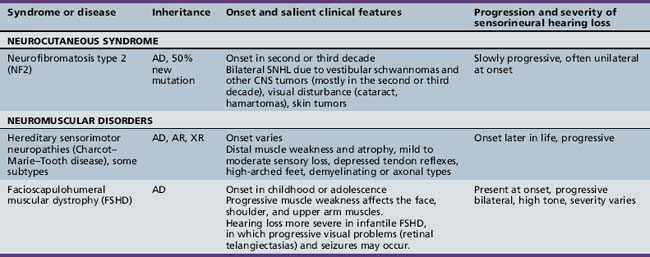
Some axonal and demyelinating auditory neuropathies occur as part of a systemic neuropathy, most often genetic, as hearing loss in acute or chronic autoimmune polyradiculoneuropathy is exceptionally rare. “True” auditory neuropathy may be a feature of some hereditary sensorimotor neuropathies (e.g., Charcot–Marie–Tooth disease) [Ouvrier et al., 2007]; Friedreich’s ataxia, where the neuropathy is the consequence of ganglion cell degeneration [Lopez-Diaz-de-Leon et al., 2003; Spoendlin, 1974]; or brain diseases with both a leukodystrophy and demyelinating neuropathy, like Cockayne’s syndrome [Rapin et al., 2006]. “True” auditory neuropathy as an entity with the particular audiologic characteristics described earlier did not come to widespread attention until it was studied systematically in a genetic isolate with a variant of hereditary sensorimotor neuropathy [Starr et al., 1996]. It may be associated with vestibular impairment [Kaga et al., 1996]. Compression of the nerve by a mass in the cerebellopontine angle or internal auditory canal is a well-known and more common cause of conduction block in the auditory nerve in adults; in children, acoustic tumors virtually always signal neurofibromatosis type 2 [Martuza and Eldridge, 1988]. As is well known, acoustic tumors start in the vestibular division of cranial nerve VIII and compress the cochlear division.
Demyelinating auditory neuropathy slows conduction; both axonal and demyelinating neuropathies desynchronize volleys. The result is delay, decreased amplitude, distortion, or abolition of the ABR, and worse speech discrimination than expected from the pure tone threshold, although, admittedly, that threshold can vary widely [Starr et al., 2004]. Auditory neuropathy can lead to deafness when severe. This has led to audiologists using “auditory dys-synchrony” as synonymous with “auditory neuropathy” [Berlin et al., 2003; Hood and Berlin, 2009], taken to mean pathology limited to the inner hair cells, their synapses with type I spiral ganglion neurons, or any part of that cell, with preservation of outer hair cell function. Audiologists’ main concern is to differentiate between inner hair cell and spiral ganglion cell pathologies, which necessitates other tests, as viable ganglion cells, but not neurons, determine eligibility for a cochlear implant. Although this generic use of the term “auditory neuropathy” may be acceptable, even in nonsyndromic cases without evidence of eighth nerve involvement, we stress that it is clearly unacceptable for cases where the pathology is in the brainstem. There are mixed auditory neuropathies and central auditory disorders, like neonatal hyperbilirubinemia, recently shown in the Gunn rat to involve not only kernicterus of the auditory and vestibular brainstem nuclei and basal ganglia, but also of the auditory nerve and spiral ganglia neurons, sparing the hair cells [Shaia et al., 2005].
Central Auditory (Retrocochlear) Hearing Loss
Brainstem hearing loss
Hearing loss attributable to dysfunction or damage to the central auditory pathway is infrequent in children and rarely gives rise to total hearing loss because of the bilaterality of central acoustic projections. Adrenoleukodystrophy may impair hearing because of demyelination of the brainstem or acoustic radiations [Grimes et al., 1983; Kaga et al., 1980]. An occasional brainstem glioma and other brainstem pathologies, such as disseminated encephalomyelitis, may impair hearing [Yagi et al., 1980]. As mentioned above, mixed loss due to involvement of peripheral and brainstem pathology includes kernicterus, neonatal hyperbilirubinemia, and compression of the acoustic nerve and brainstem by a neoplasm.
Cortical deafness
This rare cause of hearing loss implies bilateral pathology in the acoustic radiations or in the auditory cortex. Auditory sensitivity may be preserved, but recognition of the source of sounds and decoding of the acoustic code of language (phonology) are impaired. The best-documented example is acquired epileptic aphasia (Landau–Kleffner syndrome), the rare insidious loss of language in children with temporoparietal epileptiform electroencephalogram (EEG) activity with or without clinical seizures. It usually reflects verbal auditory agnosia, or, in severe cases, auditory agnosia or cortical deafness [Kaga, 1999b; Klein et al., 1995; Lewine et al., 1999].
Classification by Severity and Shape of Hearing Loss
The lower limit of normal hearing has been set at 20 dB. Hearing loss can be classified by its severity (Table 7-1). Mild flat losses of up to 40 dB do not preclude the acquisition of speech but may significantly impoverish it, the degree of language handicap depending on the age of the child and the severity and configuration of the hearing loss. If the loss is present from early infancy, it is likely to delay speech [Abraham et al., 1996; Gravel et al., 1996], especially if the child is socially deprived or otherwise handicapped [Wallace et al., 1996]. Middle ear effusion in an older child with well-established verbal skills constitutes a minor handicap. Children with even mild losses who are having academic difficulty may benefit from amplification and preferential seating in school and from a mild-gain individual or classroom FM system.
Table 7-1 Severity of Hearing Loss
| Severity* | Consequences† | Management |
|---|---|---|
| None: 0–20 | Normal speech and language | None |
| Mild: 25–40 | Speech and language normal, mildly delayed, or slightly impaired in normal children, detrimental in multiply handicapped or socially deprived children | Repeated testing Preferential seating May need speech therapy and FM loop in school |
| Moderate: 45–60 | Speech and language usually delayed and defective, especially consonants; significant impairment in most children | Hearing aid; FM loop in school Auditory training; speech and language therapy Normal school and resource room with teacher of the hard of hearing |
| Severe: 65–80 | Speech and language usually severely defective; require intensive specialized training | Hearing aid, FM loop, cochlear implant? Auditory training, sign language desirable; focus on written language Unimplanted: most do best in special school or class for the deaf, at least initially |
| Profound: 85+ | Most develop poor or no speech, even with early specialized training; language severely defective in most, who sign much better | Hearing aid, cochlear implant Intensive auditory training Sign language critically important (total communication or, better, American Sign Language); focus on written language Unimplanted or unsuccessfully implanted: most do best in specialized programs for the deaf |
* Severity is measured in decibels of hearing loss (dB HL)/pure tone average. Each category of severity is recorded in dB HL or decibels above the average normal hearing threshold. Pure tone average refers to the mean of thresholds at 500, 1000, and 2000 Hz.
† Not all consequences of hearing loss of designated severities are listed. These characteristics vary among individual children within a category.
In most children, moderate loss of 45–60 dB interferes severely with, but does not preclude altogether, the acquisition of speech. Children with moderate and severe (65–80 dB) hearing loss gain the most from hearing aids [Nozza, 1996; Roeser et al., 1995]. Virtually all children with severe hearing loss require some type of special education to learn language adequately. Profound loss (>85 dB) and total loss preclude the acquisition of language without intensive specialized education. Strictly speaking, the term deafness applies only to these last two groups, but it is often used loosely in referring to all children who cannot learn language without special assistance.
Children with very limited or no hearing benefit little from hearing aids, except, perhaps, for the awareness that a sound has occurred. Most profoundly deaf adolescents and adults do not wear hearing aids, although, as children, they should be encouraged to wear aids because it is difficult to predict who will benefit from them. Children with profound but incomplete hearing losses gain more from their cochlear implants than those with pure tone average thresholds exceeding 100 dB [Pulsifer et al., 2003], and toddlers implanted early (from 6 months to 2 years) benefit much more from their implants than children implanted in middle childhood [Govaerts et al., 2002], [Sharma et al., 2009]. As stressed earlier, functional spiral ganglion cells are required for cochlear implantation.
Classification by Age at Onset of the Hearing Loss
Age at deafening has a profound effect on its consequences. Schein and Delk, in their outstanding sociologic study of the deaf in the United States [Schein and Delk, 1974], list three main types of age-related childhood hearing loss: congenital or prelingual, perilingual, and postlingual. Prelingual means prenatal or infantile onset. Perilingual means onset during the acquisition of language and its grammatical rules, arbitrarily set by age 3 years. Most preschoolers who lose their hearing soon after age 3, i.e., technically postlingually, retain little expressive speech or language comprehension, even if great effort is made to preserve it, although they may have somewhat less difficulty reacquiring speech than children who never had hearing. Thus, age 3 years seems too young to mark the division between the prelingually/perilingually and the postlingually deaf. Children who do the worst are those who are essentially totally deaf – for example, from purulent meningitis – in whom pathology reveals total collapse of the entire endolymphatic organs, both vestibular and cochlear. Very young children need to hear to continue speaking. Prompt loss of speech is also typical of children with acquired epileptic aphasia with word deafness. Even if they are 5 years or older, they are likely to be mute and to start communicating with gestures or writing, not speech.
Pediatric deafness is mostly infantile. Schein and Delk found that the great majority of the prevocationally deaf – those whose deafness began before the age of 19 years, before they started working in a job or profession – are prelingually or perilingually deaf, proof that severe hearing loss acquired after infancy is uncommon. Any young child with severe loss of hearing from an ostensibly irreversible acquired cause like purulent meningitis (not epilepsy!), who shows no sign of improvement after several weeks, needs urgent electrophysiologic testing to find out whether at least some of the spiral ganglion cells have survived so the child can undergo prompt cochlear implantation in order to spare as much speech as possible. Another reason for early implantation is that ossification of the cochlea [Isaacson et al., 2009] and at least some trans-synaptic degeneration of the spiral ganglion neurons are long-term sequelae of purulent endolabyrinthitis which jeopardize successful implantation [Papsin and Gordon, 2007].
Classification by the Course of the Hearing Loss
Most severe sensorineural hearing loss is static, but about 10 percent, notably that caused by cytomegalovirus infection [Fowler et al., 1997] or a neurologic disease such as an acoustic tumor, and a significant proportion of genetic hearing loss (Table 7-2), is progressive [Davis et al., 1995]. Hearing loss that worsens rapidly during infancy is unlikely to be recognized as progressive, but it probably accounts for some of the children who pass neonatal screening and later turn out to be deaf. Genetic hearing loss that does not become apparent until later childhood or adult life is progressive by definition. It is now clear that both genetics and deleterious environmental influences, including acoustic trauma, contribute to the ubiquitous presbyacusis of old age. Genetic information about progressive hearing loss is generally insufficient to predict the course of a child’s hearing loss because different mutations of a given gene can result in disparate phenotypes, depending on the amount of protein synthesized. Unless the child belongs to a large family with many affected members with similar courses, who inherited an identical mutation from a distant founder, early prediction of outcome is not fully reliable. Slowly progressive hearing loss tends to denote less detrimental molecular effects or, perhaps, greater susceptibility to environmental influences than those causing early deafness. Other hearing loss, such as that due to middle ear effusion, episodic acoustic trauma, salicylate intoxication, and Ménière’s disease, fluctuates. Reasons for fluctuation are not always apparent.



Pathology
Malformations may be part of a branchial arch malformation syndrome, which may or may not be genetic, which also involves the face and, in some children, is associated with anomalies in other organ systems such as the spine, limbs, gastrointestinal tract, or genitourinary system [Toriello et al., 2004]. These associated anomalies tend to arise at the same time during gestation as the ear anomalies. Because the neural crest contributes to the formation of the ear and these other systems, the entire malformation complex may be regarded as a cristopathy. Ear anomalies may also be associated with anomalies of the posterior fossa, brainstem, other cranial nerves, eyes, and brain [Wiznitzer et al., 1987].
To recapitulate, sensorineural hearing loss often results from hair-cell or synaptic dysfunction or hair-cell loss, with or without associated damage to the neurons of the spiral ganglion [Merchant et al., 2005; Dublin, 1976] and stria vascularis. The cells of the organ of Corti and spiral ganglion are end-state cells incapable of cell division [Ruben, 1967], which explains the irreversibility of most sensorineural hearing loss. Spiral ganglion neuronal pathology leads to Wallerian degeneration in the acoustic nerve and may result in delayed transneuronal degeneration in the central auditory pathway [Ruben and Rapin, 1980; Gandolfi et al., 1984]. Primary nerve deafness or auditory neuropathy may be axonal, demyelinating, or mixed, the latter inevitable with long-standing demyelination or axonapathy; it may even be potentially reversible in compression by a tumor or an infiltrative process of the meninges.
Infections, including bacterial meningitis, congenital rubella, and cytomegalovirus; trauma; and vascular disease are unselective and are likely to damage all elements of the cochlea and vestibule; they may even result in cochlear ossification [Douglas et al., 2008]. Bacterial meningitis may deafen because of purulent endolabyrinthitis, or by lymphocytic infiltration of the vestibule and cochlea and cellular apoptosis, which suggest an immune pathophysiology [Nadol, Jr., 1997]. Some viral infections are surprisingly selective, producing only a high-tone loss rather than total destruction of the cochlea. Pathologic study of the ears of 66 patients who were profoundly deaf revealed that those with presumptive postnatal viral and bacterial labyrinthitis or with malformative and genetic causes were less likely to have residual spiral ganglion cells than those in whom deafness resulted from aminoglycoside toxicity or sudden idiopathic hearing loss [Nadol et al., 1989]. Ototoxic drugs [Stavroulaki et al., 2002; Stavroulaki et al., 2001], some other toxins, and sound trauma [Salvi et al., 1986] are highly selective as to the cell type they destroy. Also highly selective are the genetic hearing loss syndromes, which molecular biology and neuropathology are rapidly elucidating in animal models (see below).
Genetic sensorineural hearing loss is more often the result of malfunction or early death of cells in the cochlea than of their failure to form, although some of those that appear non-syndromic are associated with a variety of malformations of the bony labyrinth [Westerhof et al., 2001]. As previously stressed, which cell type is affected by genetic hearing loss varies [Bahmad et al., 2008; Cremers et al., 1998; Sampaio et al., 2004]. In the Mondini malformation, the cochlea is incompletely developed and involvement of the semicircular canals varies [Sennaroglu and Saatci, 2002]. Enlargement of the vestibular aqueduct and endolymphatic sac may be isolated or occur with other inner ear anomalies [Yang et al., 2009], and be accompanied by a variably severe or fluctuating hearing loss and, occasionally, by paroxysmal vertigo. Patency of the cochlear aqueduct may be associated with a perilymphatic or even CSF fistula into the middle ear, usually around the stapes footplate [Liu et al., 2008; Reilly and Weber, 1996], which, in some children, can cause incapacitating episodes of vertigo.
The pathology of deafness in humans lags behind its rapid progress in animal models. Examination of the ear requires sawing into the petrous bone, lengthy decalcification of the specimen, and serial sectioning. Because of the long interval between the diagnosis of hearing loss and the death of patients, physicians are unlikely to remember the details of the hearing impairment. The ear is rarely removed at postmortem examination, even though there are a number of specialized laboratories and temporal bone banks willing to study specimens, provided they are accompanied by adequate information regarding the patient’s hearing and clinical status. The National Institute of Deafness and Other Communication Disorders of the National Institutes of Health provides a listing of these laboratories, which may be obtained from <nidcdinfo@nidcd.nih.gov>.
Etiology
There are three etiologic categories of hearing loss: genetic, acquired, and unknown. Genetic hearing loss accounts for some 50–65 percent of children who fail newborn screening, and known acquired loss causes some 25–30 percent [Morton and Nance, 2006], [Wang et al., 2011], while the remainder are of unknown etiology, a proportion bound to shrink as more individuals undergo genetic and immunologic testing. An unknown number of cases may be multifactorial and account for some of the delayed or progressive apparently nongenetic cases (for example, some subclinical infections, others with mitochondrial inheritance of susceptibility to aminoglycoside antibiotics, and other toxins) [Kokotas et al., 2007].
Genetic Causes
A number of useful reviews [Petersen et al., 2008; Petersen and Willems, 2006; Petersen, 2002; Kokotas et al., 2007; Friedman and Griffith, 2003; Dror and Avraham, 2009] discuss the cell biology and pathophysiology of known genetic hearing loss, although they cannot keep up with progress in the field. Two indispensable regularly updated web resources are Online Mendelian Inheritance in Man (OMIM) (http://www.ncbi.nlm.nih.gov/omim/) and Hereditary Hearing Loss Homepage (http://hereditaryhearingloss.org). The following abbreviated review, which is limited to a few hearing loss types that child neurologists may encounter, depends heavily on these sources. The somewhat outdated book by Toriello and colleagues [Toriello et al., 2004] remains useful because of the clinical details and pictures it provides.
Genetic hearing loss is generally bilateral but may be asymmetric, and may be static or progressive. The most salient asymmetric dominant hearing loss is neurofibromatosis type 2 (NF2) [Asthagiri et al., 2009]. Mendelian gene defects can be autosomal-recessive or autosomal-dominant, X-linked recessive – even, exceptionally, Y-linked – or mitochondrial. Compound heterozygous mutations of recessive genes are generally more prevalent than homozygous mutations. Sporadic deafness may be due to unrecognized recessive disease, to new dominant mutation, cytogenetic anomaly, or single-stranded copy number variation (CNV). When insertions, deletions, translocations, or other chromosomal rearrangements are large, they may affect two or more adjacent genes, and many are detectable by high-resolution chromosome banding studies. As modern genetics keeps demonstrating, what was thought clinically to be a single disease regularly turns out to be several, resulting from mutations of distinct genes that affect a common pathophysiologic or metabolic pathway. Further, clinically distinct syndromes often arise from allelic or nonallelic mutations in different parts of the same gene, with more or less severe consequences for protein synthesis. For example, the amount of the protein pendrin determines in large measure the severity of the phenotype in Pendred’s syndrome, and in some cases whether the trait is inherited in dominant or recessive fashion. Because of these complexities, we are not giving the names of most genes we mention in the text or tables; nor do we try to mention all of the phenotypes resulting from the mutations of a particular gene. OMIM is more up to date and should be consulted for details because only super-specialists can hope to dominate the genetics of hearing loss.
Syndromic Genetic Hearing Loss
Several hundred syndromic forms account for some 30 percent of all genetic deafness [Toriello et al., 2004]. We list only a few of the more prevalent or salient ones in Table 7-2. Sex-linked disorders, both syndromic and nonsyndromic, are rare [Petersen et al., 2008]. Some syndromic losses are easy to spot because of their characteristic features, notably Treacher–Collins [Marres, 2002] and Waardenburg’s syndrome [Nance, 2003]. Deafness is not the most prominent aspect of the disorder in others, and it only becomes symptomatic later in life in still others [e.g., the “auditory neuropathy” of Friedreich’s ataxia [Lopez-Diaz-de-Leon et al., 2003], some hereditary sensorimotor neuropathies [Starr et al., 1996; Nicholson et al., 2008], and dominant optic atrophy (OPA1) [Huang et al., 2009]. Table 7-3 lists a few pediatric neurologic disorders associated with hearing loss. Among the polysaccharidoses, the loss is generally mixed, i.e., conductive and sensorineural; it is most consistent and severe in Hunter’s syndrome but is reported in the others as well [Cho et al., 2008; Riedner and Levin, 1977; Simmons et al., 2005]. Hearing loss in the leukodystrophies may be due to involvement of the central white matter – as in adrenoleukodystrophy [Pillion et al., 2006], or of both peripheral and central myelin – as in Cockayne’s syndrome [Rapin et al., 2006]. Hearing loss is a feature of a significant proportion of mitochondrial diseases [DiMauro and Schon, 2003; Finsterer et al., 2009]. As is well known, if the mutation affects the mitochondrial DNA, all the children of an affected mother are at risk for hearing loss, although not necessarily of equal severity, whereas mitochondrial deafness arising from nuclear DNA mutations follow Mendelian inheritance patterns.
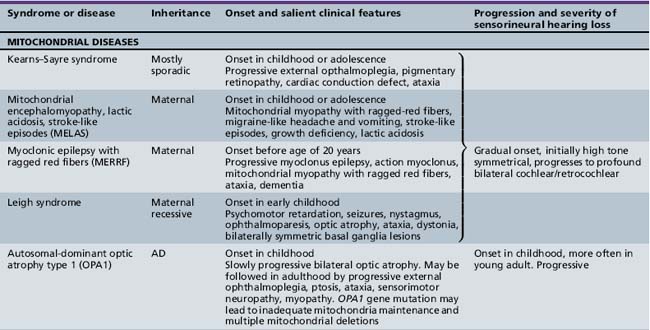
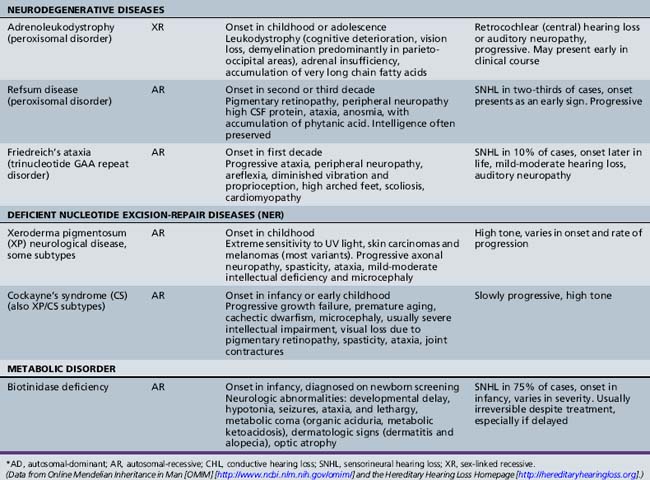
Nonsyndromic Genetic Hearing Loss
Nonsyndromic cases account for approximately 70 percent of genetic hearing loss. Close to 80 percent of prelingual nonsyndromic deafness is autosomal-recessive, 20 percent dominant, 1 percent X-linked, and less than 1 percent mitochondrial [Morton and Nance, 2006; Petersen et al., 2008; Petersen and Willems, 2006; Petersen, 2002; Kokotas et al., 2007]. Since nonsyndromic loss shares phenotypes, genetic testing is required to identify its numerous underlying genotypes. Genetic testing in the clinic tends to focus on identifying the more prevalent mutated genes, not on identifying the many specific mutations of any given one, although current technology may change this. Laboratories specialized in this work have made breathtaking progress toward understanding the function of the genes for syndromic and nonsyndromic hearing loss. The Friedman and Griffith review [Friedman and Griffith, 2003] is particularly helpful because it groups disorders by the roles of the involved genes according to the function of individual cochlear cells.
Cochlear channelopathies are responsible for a major category of nonsyndromic hearing loss. The GJB2 gene is the most common recessive gene for profound prelingual nonsyndromic deafness. It codes for connexin 26, a gap-junction protein of the many cells of the inner ear that participate in recycling K+ to the endolymph after sound excitation. Mutations for connexin 30 are rarer causes of hearing loss [Liu et al., 2009]. Connexin 31 mutations are associated with some of the hereditary sensorimotor neuropathies (HSMN), a few of which also involve hearing. Some of the other genes responsible for axonal or demyelinating HSMN and the trinucleotide repeat (GAA) gene of Friedreich’s ataxia are responsible for occasional childhood-onset progressive hearing loss [Ouvrier et al., 2007]. Another gene, KCNQ4, also involved in K+ recycling, produces a dominant progressive high-tone hearing loss in the second and third decades [Petersen, 2002]. Lack of the protein pendrin results in Pendred’s syndrome, a severe recessive prelingual hearing loss which can be syndromic or nonsyndromic, and is often associated with EVA or a Mondini malformation. EVA suggests a direct or indirect influence of impaired anionic transport of iodide and chloride on the fetal endolymph, whereas development of a euthyroid goiter is delayed until the second decade [Petersen and Willems, 2006]. The sodium/potassium ATPases play a major role in transmembrane ionic transport essential for maintaining ionic gradients in a number of cell types of the cochlea [Carelli et al., 2009]; in rats they are also involved calcium signaling [Meyer et al., 2009] and in glutamate transport at the synapses between inner hair cells and spiral ganglion type I neurons [McLean et al., 2009; Santarelli et al., 2009]. Specificity of otoferlin for release at excitatory glutamatergic inner hair-cell synapses means that this cause of profound prelingual deafness is of the “auditory neuropathy” type, because the outer hair cells and their OAEs are preserved. In contrast, mutations affecting claudin, a major structural protein of intercellular tight junctions, produce deafness by rapid selective degeneration of the outer hair cells, with only later degeneration of the inner hair cells [Petersen and Willems, 2006].
Among the many other causes of sensorineural hearing losses, there are mutations of structural genes for the tectorial membrane, genes specific to cilia, and mutations of a number of the myosin cellular motors. These motors affect bending of the hair cells in response to movement of the basilar membrane and are responsible for some subtypes of Usher’s syndrome, other subtypes of which are due to inadequate adhesion molecules (cadherins). Cellular motors require energy provided by the adenosine triphosphate (ATP) generated by the mitochondrial electron transport chain [Kokotas et al., 2007]. The high prevalence of hearing loss in mitochondrial diseases is most likely linked to inadequate generation of the energy required not only for the motors but also to maintain ionic gradients for continuous fast neurotransmission of sound. Mitochondrial function plays a role in ototoxicity, acoustic trauma, and the effects of anoxia. Mutation of the dominant nuclear mitochondrial gene OPA1 causes early optic atrophy with later progressive hearing loss and ataxia of variable severity. Other mitochondrial diseases associated with hearing loss are listed in Table 7-3. This short list is but an illustration of the complexities of the ever-growing number of potential molecular mechanisms responsible for hearing loss.
Malformations
Ear malformations may be sporadic or genetic, and involve one or several parts of the ear, some isolated, some part of complex malformation syndromes. Bilateral anomalies are more likely than unilateral ones to involve the brain and other organ systems. Some, but by no means all, are associated with intellectual deficits or autism [Wiznitzer et al., 1987]. The severity of malformations of the external ear and face does not predict outcome reliably unless associated with a major brain abnormality, so that a reliable prognosis cannot be provided in infancy.
Stenosis or atresia of the external ear canal may occur in isolation or be associated with malformation of the pinna and middle ear, and, in some children, the inner ear [Toriello et al., 2004]. Atresia produces a severe conductive loss and requires neonatal referral for bone conduction aids. The pinna may be absent, reduced to a small remnant (microtia), associated with one or more preauricular tags, or be present but malformed. The pinna with its lower half missing or deformed strongly suggests the CHARGE association (i.e., coloboma, heart defects, atresia choanae, retardation of growth and development, genitourinary problems, and ear anomalies) [Stromland et al., 2005]. Malformations of the ossicular chain and cochlea, and those of the central auditory canal containing cranial nerves VII and VIII are best visualized with ultra-thin, high-resolution computed tomography (CT) of the temporal bones. Patency of the cochlear scala tympani is critical for implantation candidacy. Microtia is frequently associated with ipsilateral facial palsy or deficient growth of the face (hemifacial microsomia), and conductive or mixed hearing loss [Carvalho et al., 1999]. In Möbius’ syndrome sensorineural hearing loss and involvement of other cranial nerves besides VI and VII may occur [Griz et al., 2007]. As previously mentioned, some cochlear anomalies are associated with perilymphatic/CSF fistulas into the middle ear, usually through the stapes footplate; they may produce fluctuating or progressive hearing loss and severe intermittent labyrinthine dysfunction with vertigo, nausea, or vomiting, and they may provide a path for a purulent endolabyrinthitis or meningitis [Reilly and Weber, 1996].












