Chapter 85 Cerebrovascular Disease in Children
Overview and Definitions
It is important to distinguish stroke from similar neurological disorders that are discussed elsewhere. Perinatal strokes, including AIS, CSVT, and other varieties, are a common and important variety of cerebrovascular disease discussed in detail in Chapter18. Global hypoxic-ischemic brain injury (HIE) is also common in newborns and children. While the mechanism of cellular injury is similar, these lack the isolated focal distribution of injury that defines vaso-occlusive stroke. Therefore, global HIE diseases in preterm and term neonates (Chapter 17,19), as well as global and watershed HIE (Chapter 17), are covered elsewhere. Specific patterns of intracranial hemorrhage, including germinal matrix bleeding unique to the premature brain (Chapter 19) and traumatic extra-axial intracranial hemorrhage (Chapter 18), also are covered elsewhere. The many conditions that may mimic childhood stroke clinically are reviewed below, with cross-references to corresponding chapters.
The primary objective of this chapter is to provide the reader with a comprehensive, clinically relevant review of the elements of cerebrovascular disease encountered in infants and children. Recent general review articles are available [Pappachan and Kirkham, 2008; Dlamini and Kirkham, 2009; Amlie-Lefond et al., 2008], and section-specific review articles and consensus-based guidelines are referenced below. The information presented is based on the best available evidence to the end of 2009. In order to highlight the limited level of evidence in many areas, relative standards of evidence criteria [Cook et al., 1995] are provided where appropriate. With almost no randomized clinical trails and limited prospective studies, many studies are classified as level IV or V (retrospective case series). As evidence is evolving rapidly, the reader is encouraged to supplement this chapter with searching the current literature regarding specific topics from 2010 on.
Epidemiology of Pediatric Stroke
Stroke is commonly encountered in the practice of pediatric neurology. Hospital administrative data suggest that cerebrovascular disorders are among the top ten causes of death in children, with rates highest in the first year of life [Murphy, 2000]. Stroke-related mortality in young children may have decreased in the past two decades [Fullerton et al., 2003b]. The importance of “stroke in the young” (i.e., <40 years of age) is recognized increasingly and children account for nearly one-quarter of such cases [Kerr et al., 1993]. The burden of pediatric stroke is amplified by the duration of the adverse neurological outcomes suffered by most children, lasting decades. Economic costs of childhood stroke are substantial in the acute [Janjua et al., 2007; Perkins et al., 2009] and medium term [Lo et al., 2008a]. Less studied are the long-term costs, which are likely greater [Perkins et al., 2009]. The life-long stroke-attributable socioeconomic burden per affected individual, therefore, is substantial, and comparable in many regards to adult stroke [Taylor et al., 1996], to which approximately 3 percent of health-care expenditures in developed countries currently is dedicated [Evers et al., 2004]. The burden of pediatric stroke appears global, with recent international research initiatives further defining regional epidemiology [International Pediatric Stroke Study Web-site, 2010].
In the past three decades, incidence rates for pediatric stroke appear to be increasing. Updated summaries of available epidemiological data to date, including comparisons of ischemic versus hemorrhagic stroke, are available [Mallick and O’Callaghan, 2009a; Lynch et al., 2002]. While nearly 20 publications have examined stroke incidence rates, comparability is compromised by differences in methodology, definitions, and inclusion criteria. Factors such as age range (neonates or not), ischemic stroke definitions (CSVT included or not), and hemorrhage (included or not) account for variability in quoted rates. Many studies have relied on health-care administration databases and international classification of disease codes, with very few studies employing true population-based methodology with direct diagnostic classification [Agrawal et al., 2009].
Previously reported combined rates for ischemic and hemorrhagic pediatric stroke in the United States were 2.5–2.7 per 100,000 children per year [Broderick et al., 1993; Schoenberg et al., 1978]. These values are consistent with more recent studies [Fullerton et al., 2003b], including one population-based study using imaging-confirmed diagnoses [Agrawal et al., 2009]. A prospective, population-based Canadian registry estimated total ischemic stroke (AIS and CSVT) incidence at 2.2 per 100,000, including 1.8 for AIS and 0.4 for CSVT (unpublished data). Serial calculation of pediatric stroke rates across the decade ending in 1999 demonstrated a near-doubling from 2.8 to 5.4 per 100,000 [Kleindorfer et al., 2006]. Although less valid rigorous diagnostic criteria were used, even higher rates were reported in the National Hospital Discharge Survey (10.7 per 100,000) [Lynch et al., 2002] and in a truly population-based regional European study (13 per 100,000) [Giroud et al., 1995]. In Victoria, Australia, the incidence of childhood CSVT was estimated at 0.34 per 100,000 children per year [Barnes et al., 2004]. The reported relative proportions of hemorrhagic vs. ischemic stroke in childhood are inconsistent across multiple studies. Regarding which type of stroke predominates, some report hemorrhagic [Broderick et al., 1993; Schoenberg et al., 1978], others ischemic [Lanthier et al., 2000; Lynch et al., 2002; Giroud et al., 1995], and still others, neither [Fullerton et al., 2003b; Earley et al., 1998].
At least two factors have increased the number of diagnosed childhood strokes. First, widespread availability of more sensitive diagnostic tests, particularly magnetic resonance imaging (MRI), has increased [Wiznitzer and Masaryk, 1991; Venkataraman et al., 2004; Warach and Baron, 2004]. Second, more effective treatments have increased survival in previously lethal pediatric diseases predisposing to stroke, including prematurity, congenital heart disease, sickle cell disease, and childhood cancers. Despite improved recognition, ischemic strokes likely remain under-diagnosed, particularly CSVT. A high degree of clinical suspicion and appropriate use of diagnostic tests remains essential.
Arterial Ischemic Stroke
Pathophysiology
Arterial Circulation: Anatomy and Vascular Patterns
Understanding the pathophysiology, clinical presentation, possible etiologies, and potential outcomes of childhood AIS requires an understanding of craniocervical arterial circulation. The brain receives arterial blood via two principal systems: the “anterior” circulation, consisting of the paired internal carotid arteries, and the “posterior” circulation, consisting of the paired vertebral arteries that join to form the basilar artery (“vertebrobasilar system”) (Figures 85-1 to 85-3). The anterior and posterior communicating arteries link these systems to form the circle of Willis. The major cerebral arteries arising from the circle of Willis are the paired anterior, middle, and posterior cerebral arteries. The anterior and middle cerebral arteries are the major branches of the internal carotid artery, and the posterior cerebral arteries are the major branches of the vertebrobasilar system. Small “perforating arteries” arise from these proximal vessels to supply deep brain regions, such as the basal ganglia and thalamus. These include lenticulostriate branches from the anterior circulation, and thalamostriate vessels from the posterior vessels. Circulation to the posterior fossa includes perforating arteries into the midline of brainstem, and major circumferential arteries supplying the lateral brainstem and cerebellum (the posterior inferior, anterior inferior, and superior cerebellar arteries). Inter-individual variability in the anatomy of the circle of Willis is frequent and can be relevant clinically. Anastomoses, both proximally at the circle of Willis and distally through smaller leptomeningeal vessels, can provide collateral blood flow, the degree of which significantly affects the territory of brain perfused by a major cerebral artery.
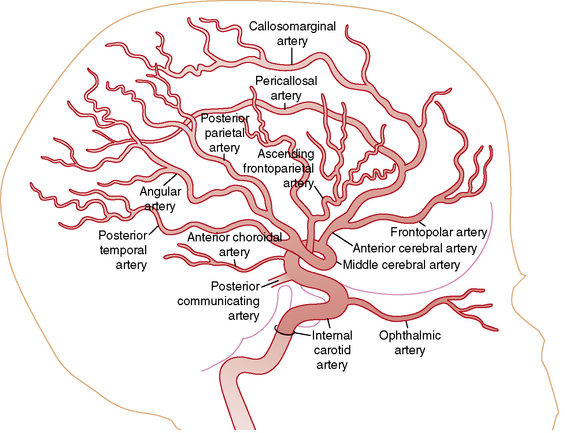
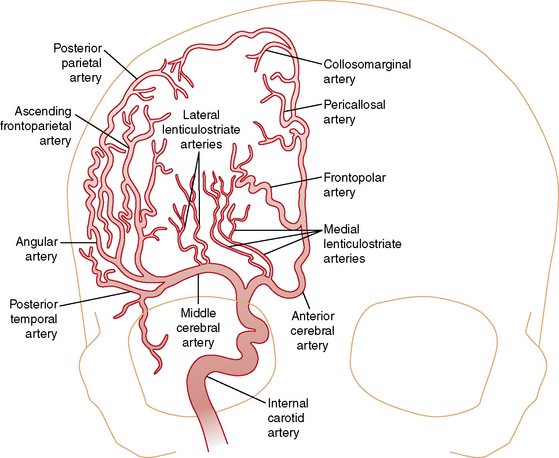
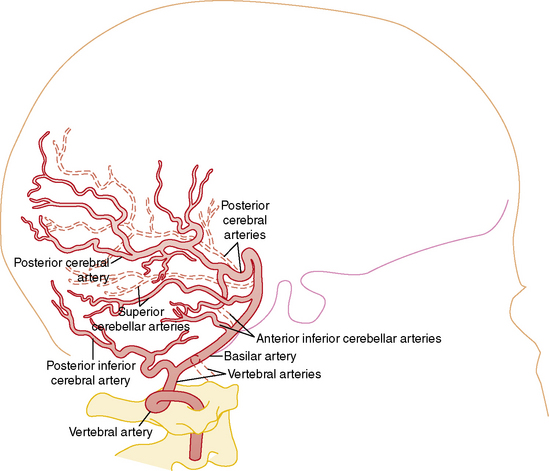
Vascular patterns of AIS can be classified as large- or small-vessel infarcts. Infarction due to occlusion of large-caliber cerebral arteries usually results in peripheral wedge-shaped lesions involving the cerebral cortex and subjacent white matter in characteristic distributions (Figure 85-4). Most AIS involves the middle cerebral artery (MCA), which, like the other major vessels, courses to the cerebral surface where pial branches supply the cortex and underlying white matter. AIS often occurs in recognizable patterns within the MCA territory, including:
As end-arteries, lenticulostriate lesions lack collaterals, conferring a higher risk of permanent infarction. Despite being small, such lesions also tend to affect critical structures and functional tracts, such as the internal capsule (Figures 85-5 and 85-6). Such specific recognizable patterns also help predict the underlying etiology, with isolated lenticulostriate AIS commonly associated with parainfectious or inflammatory arteriopathy [Askalan et al., 2001; Chabrier et al., 1998; Braun et al., 2009]. In population-based studies, such small-vessel-distribution infarcts account for 50 percent of childhood AIS. For unknown reasons, AIS more frequently affects the left hemisphere [deVeber, 2000].
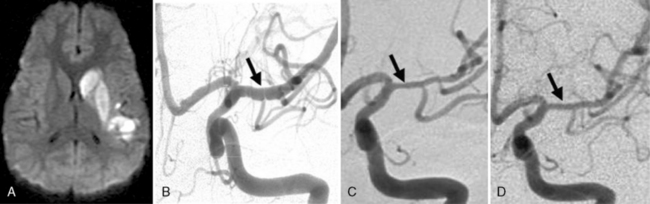
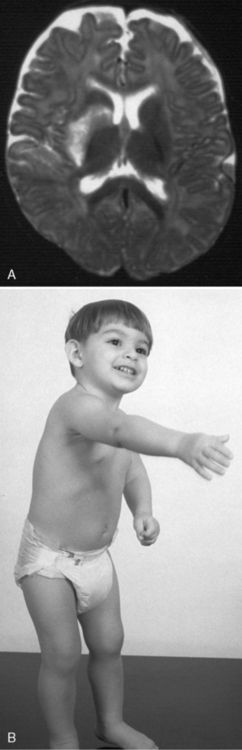
Fig. 85-6 Small-vessel arterial infarct of unknown etiology in a 10-month-old male.
(Courtesy of Division of Neuroradiology, Hospital for Sick Children, Toronto, Ontario, Canada.)
Mechanisms of Thromboembolism
Thrombosis is a product of the coagulation and platelet hemostatic systems. In situations of blood stasis or slow flow, such as the venous system, the coagulation system predominates, resulting in fibrin-rich thrombi. However, in arteries, platelet activation with platelet-rich thrombi is a significant contributor to thrombus formation, combining with coagulation activation. The balance of which system predominates varies with flow rate, shear stresses, endothelial integrity, concentrations of circulating anticoagulants (e.g., protein C and S), and other factors. Under physiologic circumstances, arterial endothelium provides an anticoagulant surface, helping to maintain blood in a fluid phase. When the arterial wall is damaged by disease or injury, thrombosis may develop through several mechanisms. Exposure of blood to arterial wall inflammation (e.g., vasculitis) or disruption of the endothelium with exposure to collagen and tissue factor (e.g., dissection) activates both platelets and fibrin formation. In situations of blood stasis or slow blood flow, including arterial occlusion or severe stenosis, the coagulation system may predominate. In contrast, rapid arterial blood flow with high “shear” forces favors platelet activation. Therefore, coagulation- and platelet-mediated thrombotic processes likely coexist in many children with AIS, the relative balance depending on the specific disease. Important developmental differences in both systems through early infancy and childhood are likely also important [Andrew et al., 1990; Carcao et al., 1998; Israels et al., 2001; Roschitz et al., 2001; Monagle, 2008a].
Mechanisms of Infarction and Developmental Brain Injury
Ischemic neuronal dysfunction can be reversible initially, but will shift to irreversible infarction with increasing duration and/or degree of ischemia or increased rates of neuronal metabolic activity. In a transient ischemic attack (TIA), no permanent parenchymal lesion responsible for the symptoms is seen on MRI [Albers et al., 2002]. This is consistent with the transient symptomatology (resolution typically within 60 minutes) and reversible ischemic neuronal dysfunction without permanent damage. In contrast, AIS clinical deficits persist and are accompanied by radiographic confirmation of infarction. Despite this logical clinical-radiographic correlation, pathological processes at the tissue level are certainly more complex and dependent on a number of factors. In AIS, a marked decrease in focal cerebral perfusion results in a central core zone of irreversibly damaged brain. Surrounding this is a less severely compromised penumbral zone where tissue is partially injured but remains potentially viable [Schlaug et al., 1999]. In the penumbra, clinical factors that increase the discrepancy between cerebral metabolic rate and the delivery of oxygen and glucose result in additional tissue injury and cell death. Secondary insults, including seizures and alterations in temperature, blood pressure, and serum glucose, increase this discrepancy between energy supply and demand. The primary objective of acute neuroprotective and fibrinolytic stroke treatment is to rescue this “at-risk” penumbral tissue to salvage functional brain.
Focal brain ischemia results in regional hypoxia and depletion of high-energy compounds (adenosine triphosphate) and carbohydrate stores. Compared to other tissues, the brain has relatively higher baseline metabolic rates and fewer energy storage systems [Trescher, 1992]. The cerebral metabolic rate for oxygen is 3.5 mL/100 g/min and, with virtually no oxygen reserve available, neuronal dysfunction results quickly when supply ceases. Brain glucose reserve is greater, allowing tissue survival up to 90 minutes with adequate oxygenation. The newborn can use lactate as a substrate for the production of energy but this capability is lost quickly. When regional hypoxia develops during ischemia, there is a shift from oxidative metabolism to glycolysis. Glucose is metabolized to lactate, and accumulating acidosis further exacerbates injury. Final infarct volume is determined in part by the balance between the rate of delivery of oxygen and glucose, and regional metabolic brain activity, particularly in the penumbra.
Several elements of pediatric cerebral hemodynamics are relevant in AIS. Cerebral perfusion is a direct function of the mean arterial pressure, intracranial pressure, and vascular resistance, and is age-related [Volpe, 2001a]. Adult cerebral blood flow (CBF) is 50 mL/100 g brain tissue/minute. Neonatal CBF is approximately 40 percent of this, while children under 3 years average 30–50 mL/mg/min. Children of 3–10 years have high CBF (approximately 100 mL/mg/min) that returns toward adult values by the end of adolescence [Altman et al., 1988]. CBF represents 10–20 percent of cardiac output in early infancy, peaks at over 50 percent by 2–4 years, and stabilizes at approximately 15 percent by 8 years [Wintermark et al., 2004]. Cerebral autoregulation refers to homeostatic regional adjustments in cerebral perfusion accomplished through caliber changes in cerebral arteries in response to alterations in perfusion pressure, carbon dioxide levels, and other metabolic factors. Developmental changes in cerebral autoregulation are recognized, though not well studied in AIS. Autoregulation is present across normal blood pressures in term newborns; however, it is impaired in preterm neonates [Volpe, 2001a]. Further maturation of cerebral autoregulatory systems occurs through at least adolescence [Vavilala et al., 2005]. Disturbed cerebrovascular autoregulation surrounding an evolving infarct may further contribute to a mismatch between cerebral metabolic demands and substrate delivery.
Neonatal and juvenile animal stroke models are providing exciting basic science knowledge regarding the cellular pathophysiology of ischemic stroke, including age-related processes distinct from adult stroke [Hou and MacManus, 2002]. Details of the neurotoxic cascade initiated by acute focal hypoxia-ischemia continue to evolve. In AIS, cell death proceeds over time by two primary mechanisms. Acute necrosis occurs over hours, whereas apoptosis (delayed cell death) occurs over days and, in neonates, likely over weeks. More detailed discussion of cell death mechanisms in the brain is found in Chapter 14. Accumulation of extracellular glutamate and activation of glutamate receptors play a major initiating role in this cascade [Aarts et al., 2002]. Accumulation of excess cytosolic calcium results in activation of pathological calcium-mediated intracellular events. The role of nitric oxide and free-radical generation, with resulting damage to critical intracellular components, is also likely important [Wang and Shuaib, 2007]. Complex neuroinflammatory processes have also emerged as potential targets for stroke neuroprotection [Shah et al., 2009].
Pharmacological and other neuroprotective strategies designed to interrupt this multifaceted ischemic cascade in stroke continue to be developed. Unfortunately, none has been successful in human stroke clinical trials, and no such trials have involved children. Hypothermia has shown benefit in the management of neonatal global HIE [Wyatt et al., 2007], but not childhood traumatic brain injury [Hutchison et al., 2008], is unproven in adult stroke [van der Worp et al., 2010] and untested in childhood stroke. Future animal models of stroke in the developing brain will be required to understand the unique aspects of these neuroprotective mechanisms in childhood AIS. Ideally, juvenile animal stroke models would incorporate features relevant to childhood AIS, including inflammatory cerebral arteriopathy, age-dependent coagulation systems and disorders, and inclusion of pediatric age-appropriate outcomes, including achievement of new learning, rather than just relearning previously acquired skills. Since the infant brain relies on programmed cell death for normal development, infant animal models studying anti-apoptotic agents will need to evaluate brain development after treatment. Translation of such results into clinical trials in the pediatric population will be challenging.
Ever since Kennard described better outcomes in younger monkeys following focal brain injury [Kennard, 1936], neuroscientists have sought to understand developmental neuroplasticity and harvest its benefits. Multiple subsequent animal models have confirmed evolving susceptibilities and potentials for recovery across the age spectrum that are dependent on many factors [Yager and Thornhill, 1997; Kolb et al., 2000; Kolb and Cioe, 2003]. AIS represents focal injury in an otherwise healthy brain, providing an ideal model for studies of plasticity. Knowledge regarding plastic changes in the brain following adult stroke has grown exponentially in the past decade [Murphy and Corbett, 2009]. Similar studies have not been performed widely in children. However, the widely held belief that increased plasticity confers an advantage to recovery after brain injury in childhood compared to adults remains unproven, and most children with AIS suffer adverse long-term outcomes. Childhood AIS outcome studies correlating age and recovery show mixed results [Bates et al., 1999; Goodman and Yude, 1996b; Westmacott et al., 2007b, 2009; Max, 2004a; Lansing et al., 2004] Fortunately, modern neurotechnologies, such as functional MRI (fMRI) [Heller et al., 2005], diffusion tensor imaging [Seghier et al., 2004], and transcranial magnetic stimulation [Frye et al., 2008], allow new windows into the study of brain plasticity in childhood AIS. Combining these basic and applied neuroscience methods in children with AIS will be critical to the development of improved interventions and better outcomes.
Clinical Features and Diagnostic Delays
The diagnosis of stroke in children is frequently delayed or missed. Multiple studies have confirmed extended intervals to diagnosis in AIS, often beyond 24 hours from symptom onset [Gabis et al., 2002; Rafay et al., 2009; Hartman et al., 2009; McGlennan and Ganesan, 2008; Srinivasan et al., 2009]. Affected children typically are brought to medical attention quickly with subsequent prolonged delays occurring in the hospital [Rafay et al., 2009]. Diagnostic delays likely reflect a combination of lack of awareness by primary care pediatric physicians, confusing neurological presentations, distracting signs and symptoms leading to erroneous lines of investigation, and a complex differential diagnosis. A false-negative rate for initial computed tomography (CT) scanning of 60–80 percent also contributes to missed and delayed diagnosis of childhood AIS. Yet prompt diagnosis is essential to afford opportunities for emergent neuroprotective management, and interventions to promote recanalization and prevent early recurrence. A high clinical suspicion, coupled with a systematic diagnostic approach, is required for timely diagnosis.
The acute onset of a focal neurological deficit in a child is stroke until proven otherwise. Acute hemiparesis is the most common presenting focal deficit but may be attributed erroneously to other causes that can mimic stroke (migraine, seizure or Todd’s paresis, encephalitis/meningitis, or demyelination) [Shellhaas et al., 2006]. Additional common focal neurological deficits include diplopia, hemianopsia, impaired gaze, dysarthria, vertigo, nystagmus, dysphasia (expressive, receptive, mixed), ataxia, hemisensory symptoms, and neglect. Seizures at the onset of stroke are also frequent in children [Delsing et al., 2001] compared with adults [Trescher, 1992; Yang et al., 1995]. In addition to focal neurological deficits, diffuse or nonfocal neurological signs and symptoms, such as headache, confusion, irritability, and behavioral changes, are frequent, especially in younger children [Trescher, 1992]. Neurological deficits can be subtle, especially in infants who are unable to manifest language deficits or verbalize subjective complaints. Careful consideration of clinical and imaging information is required to distinguish true stroke from the lengthy list of stroke mimics summarized in Table 85-1
Table 85-1 Differential Diagnosis of Strokelike Episodes in Children
| Disease | Clinical Distinction from Stroke | Imaging Distinction from Stroke |
|---|---|---|
| Migraine | Evolving or “marching” symptoms, short duration, complete resolution, headache, personal or family history of migraine | Usually normal. Migrainous infarction is rare |
| Seizure | Positive > negative symptoms. Todd’s paralysis occurs after seizure. Altered level of consciousness | May identify source of focal seizures |
| Infection | Fever, encephalopathy, gradual onset, meningismus | Markers of encephalitis/cerebritis, typically diffuse and bilateral. AIS and CSVT occur in bacterial meningitis |
| Demyelination (ADEM, MS) | Gradual onset, multifocal symptoms, encephalopathy. Past history of DM events (optic neuritis, transverse myelitis) | Multifocal lesions, typical appearance (e.g., patchy in ADEM, ovoid in MS), typical locations (e.g., pericallosal in MS), less likely to show restricted diffusion |
| Hypoglycemia | Risk factor (e.g., insulin therapy), related to meals, additional neuroglycopenic symptoms | Bilateral, symmetrical. May see restricted diffusion. Posterior dominant pattern (neonates) |
| Watershed HIE | Risk factor (e.g., hypotension, sepsis), bilateral deficits | Bilateral, symmetrical, restricted diffusion in arterial border zones |
| Inborn errors of metabolism (MELAS, mito) | Pre-existing delays/regression, multisystem disease, abnormal biochemical profiles | Possible restricted diffusion but often bilateral, not within arterial territories. MR spectroscopy (e.g., high lactate) |
| Hypertensive encephalopathy (PRES) | Documented hypertension, bilateral visual symptoms, encephalopathy | Posterior dominant, bilateral, patchy lesions involving gray and white matter, usually no restricted diffusion |
| Vestibulopathy | Symptoms limited to vertigo. Positional. Nystagmus | Normal |
| Acute cerebellar ataxia | Gradual onset, bilateral, symmetric ataxia, postviral | Normal or diffusely swollen cerebellum |
| Channelopathies (AHC, EA, FHM) | Syndromic cluster of recurring symptoms. Switching sides. Headaches. Family history | Usually normal. Need MR angiography to exclude moyamoya in AHC |
ADEM, acute disseminated encephalomyelitis; AHC, alternating hemiplegia of childhood; AIS, arterial ischemic stroke; CSVT, cerebral sinovenous thrombosis; DM, demyelination; EA, episodic ataxia; FHM, familial hemiplegic migraine; HIE, hypoxic-ischemic encephalopathy; MELAS, mitochondrial encephalopathy with lactic acidosis and strokelike episodes; MR, magnetic resonance; MS, multiple sclerosis; PRES, posterior reversible encephalopathy syndrome.
Stroke-induced deficits usually begin suddenly and severity is maximal at the onset, aiding differentiation from mimics such as migraine and demyelination. However, symptoms can also have a fluctuating or stuttering onset [Braun et al., 2007]. TIAs are not well studied in children but likely occur prior to AIS in many cases. Previously, the definition of TIA was based on a deficit duration of less than 24 hours. This has now been updated to include negative imaging, since many ischemic episodes of less than 24 hours’ duration do cause acute infarcts seen on MRI. TIA, therefore, is best defined as “a brief episode of neurological dysfunction caused by focal brain or retinal ischemia, with clinical symptoms typically lasting less than one hour, and without imaging (MRI) evidence of acute infarction” [Albers et al., 2002]. Differentiation of TIA from other causes of brief neurological deficits can be challenging. It is essential to recognize TIA symptoms, as prompt treatment may prevent a major stroke.
Risk Factors
The potential causes of AIS in infants and children are myriad, and frequently multiple in individual cases. Causes are also frequently referred to as “risk factors,” reflecting the variable strength of evidence for causality. Case-control and large population-based studies are continuing to provide important information regarding risk factors for childhood stroke. In the following discussion, we have taken an evidence-based approach that highlights well-established risk factors for childhood AIS, followed by a briefer review of additional considerations. Risk factors for perinatal stroke are distinct and discussed elsewhere (see Chapter 18). It is critical in individual children to determine all AIS mechanisms in a timely and thorough manner in order to identify risk of stroke recurrence and determine potentially appropriate therapies [Lanthier et al., 2000; Chabrier et al., 2000; Fullerton et al., 2007b]. This requires a tailored approach to investigations, based on the list of possible etiologies in Table 85-2.
Table 85-2 Potential Risk Factors for Childhood Arterial Ischemic Stroke
| Category | Common/Highly Probable | Uncommon/Possible/Uncertain |
|---|---|---|
| Arteriopathy | Inflammatory/para-infectious cPACNS (nonprogressive or progressive; large-vessel or small-vessel) Transient cerebral arteriopathy Focal cerebral arteriopathy Post-varicella angiopathy Infectious Bacterial meningitis Human immunodeficiency virus Dissection Internal carotid artery Vertebral artery Intracranial arteries Moyamoya Idiopathic Syndrome Neurofibromatosis-1, trisomy 21, other | Secondary CNS vasculitis SLE, PAN, IBD Takayasu’s arteritis Infectious: mycoplasma, toxoplasmosis, RMSF, Lyme disease, cryptococcosis, chlamydia, Japanese encephalitis, coxsackie B4 and A9, influenza A, enterovirus, parvovirus B19 Postradiation vasculopathy RSCV (Call–Flemming syndrome) Genetic: COL4A1 CT (connective tissue) disease (Marfan’s, Ehlers–Danlos) Pseudoxanthoma elasticum Congenital: PHACES, progeria, Alagille’s syndrome, dwarfism (MOPDII), fibromuscular dysplasias |
| Cardiac | Complex congenital heart disease Cardiac surgery (e.g., Fontan) Cardiac catheterization (e.g., BAS) Bacterial endocarditis Atrial septal aneurysm Atrial septal defect? Patent foramen ovale? Venous thrombosis + right-to-left shunt | Cardiomyopathy, myocarditis Aortic coarctation Severe ventricular dysfunction Atrial myxoma Valvular disease (e.g., rheumatic fever) Arrhythmia (atrial fibrillation) ECMO Cerebral angiography Embolism (air, fat, amniotic fluid) |
| Prothrombotic | Factor V Leiden Prothrombin gene 20210A Elevated lipoprotein (a) Protein C deficiency Lupus anticoagulant Anticardiolipin antibodies | MTHFR, hyperhomocysteinemia Protein S deficiency Antithrombin III deficiency Factor VII/IX/XI? Plasminogen deficiency? Dysfibrinogenemia? Sticky platelet syndrome Pregnancy, puerperium |
| Hematological | Sickle cell disease Iron deficiency anemia | Leukemia Thalassemias? Thrombocytosis? Polycythemia? Hemolytic uremic syndrome Immune thrombocytopenic purpura Thrombotic thrombocytopenic purpura |
| Medications/drugs | Oral contraceptives Chemotherapy (l-asparaginase) | Cocaine, methamphetamine, ecstasy Ergots, triptans |
| Other | Migraine Inborn errors of metabolism: Fabry’s disease, homocystinuria, mitochondrial | Metabolic syndrome: hypertension, diabetes, insulin resistance, dyslipidemia, atherosclerosis Cigarette smoking, secondhand smoke |
BAS, balloon atrial septostomy; cPACNS, childhood primary angiitis of the central nervous system; ECMO, extracorporeal membrane oxygenation; IBD, inflammatory bowel disease; MOPDII, Majewski osteodysplastic primordial dwarfism II; MTHFR, methylene tetrahydrofolate reductase; PAN, polyarteritis nodosum; PHACES, posterior fossa abnormalities, hemangiomas, arterial cervicocerebral anomalies, cardiac defects, eye anomalies, sternal defects and/or supraumbilical raphe; RMSF, Rocky Mountain spotted fever; RSCV, reversible segmental cerebral vasoconstriction; SLE, systemic lupus erythematosus.
A substantial proportion of children with AIS remain idiopathic, despite exhaustive investigations. The reported frequency of identifiable risk factors in children with AIS varies widely, depending on the population of children, criteria for risk factors, and extensiveness of etiological investigations. In single-center, tertiary-care studies, one possible risk factor was definable in 80–90 percent of children [Chabrier et al., 2000; Lanthier et al., 2000; Ganesan et al., 2003; deVeber, 2000]. Chronic diseases of childhood frequently underlie childhood AIS; however, approximately half of children are previously healthy [Ganesan et al., 2003]. Multiple risk factors converge in AIS in more than 50 percent of children, emphasizing the need for thorough investigations. Causes for AIS in children differ significantly from those in adults, in whom atrial fibrillation and atherosclerosis (associated with hypertension, dyslipidemia, smoking, and diabetes) predominate [deVeber, 2003]. A comprehensive list of potential risk factors for childhood AIS is included in Table 85-2. However, three main categories of risk factors have emerged as most prominent: arteriopathy, cardiac, and prothrombotic/hematological. These are discussed below, followed by an overview of additional risk factor considerations.
Arteriopathies
Diseases that directly affect the arteries supplying the brain have emerged as the leading cause of childhood AIS and are associated with an increased risk of both recurrence and poor outcome [Goldenberg et al., 2009; Fullerton et al., 2007b]. Recent studies have demonstrated consistently that arteriopathy is present in more than 50 percent of childhood AIS [Chabrier et al., 2000; Ganesan et al., 2003; Fullerton et al., 2007b; Amlie-Lefond et al., 2009a; Sebire et al., 2006; Braun et al., 2009]. Since arteriopathy is likely the single strongest predictor of stroke recurrence, thorough vascular imaging is essential for accurate detection and classification of arteriopathy [Lanthier et al., 2004; Strater et al., 2002; Askalan et al., 2001; Fullerton et al., 2007b; Braun et al., 2009]. Surveillance for a progressive nature in some arteriopathies is important, as it may dictate specific treatment [Danchaivijitr et al., 2006a; Kirkham, 2006; Braun et al., 2009].
Different systems have been proposed to classify childhood cerebral arteriopathies [Sebire et al., 2004], although some disagreement about terminology and proposed underlying pathophysiological processes still exists among experts. The three most important categories are discussed below:
Other, more rare, specific genetic cerebral arteriopathies include PHACES syndrome [Heyer et al., 2008], Alagille’s syndrome [Emerick et al., 2005], progeria [Merideth et al., 2008], primordial dwarfisms [Brancati et al., 2005], childhood forms of fibromuscular dysplasia [Kirton et al., 2006], and dominantly inherited COL4A1 mutations [van der Knaap et al., 2006].
Infectious/inflammatory arteriopathy
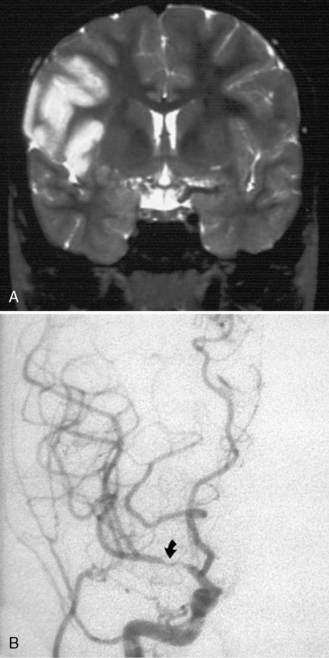
Many descriptions of this condition have been published under the name “transient cerebral arteriopathy” (TCA) [Chabrier et al., 1998; Shirane et al., 1992; Sebire, 2006] An example of the angiographic appearance of TCA is shown in Figure 85-4. Studies have suggested that such intracranial unilateral arteriopathy, even with features suggesting vasculitis on angiography, are non-progressive in more than 90 percent of cases [Braun et al., 2009]. When serial imaging demonstrates increased stenosis or extension to new arterial segments 6 months after onset, a chronic or progressive vasculitis or unilateral moyamoya may be suspected [Sebire, 2004]. Arterial occlusion, moyamoya vessels, and ACA (anterior cerebral artery) involvement may be associated with a progressive course.
Despite being very common, the exact cause or causes of Trasient cerebral Arteriopathy remain undetermined in most cases. This has resulted in the emergence of inconsistent terminology, diagnostic criteria, and treatment considerations; the debate continues. Recent efforts to acknowledge this uncertainty have used the more general term of “focal cerebral arteriopathy” (FCA) for any stenosing arteriopathy without evidence of a well-established arteriopathy, such as moyamoya or dissection [Amlie-Lefond et al., 2009a]. The most consistently suggested etiologies for TCA include infectious, postinfectious, and/or inflammatory mechanisms [Chabrier et al., 1998; Shirane et al., 1992]. TCA is considered by some to be a form of focal and self-limited vasculitis, and the term “nonprogressive childhood primary angiitis of the central nervous system” (cPACNS) has been applied [Elbers and Benseler, 2008]. However, similarities to postvaricella angiopathy (see below) [Askalan et al., 2001] have led to suspicion of parainfectious mechanisms. Still other possibilities, such as intracranial dissection, are often impossible to exclude.
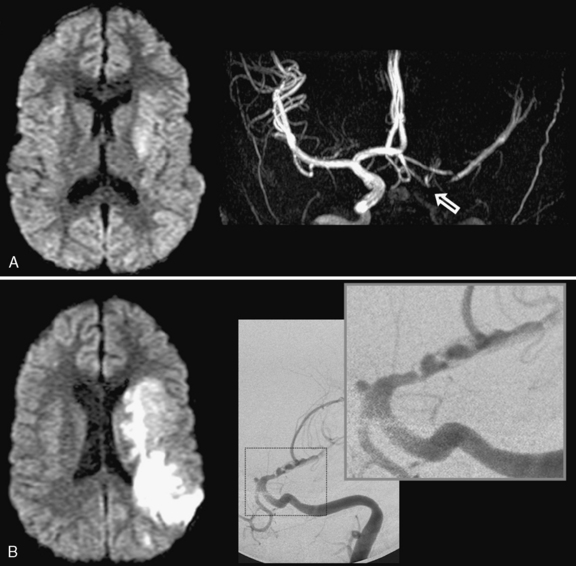
The role of infection in AIS is recognized increasingly [Emsley and Hopkins, 2008; Amlie-Lefond et al., 2009a]. The strongest example in childhood AIS is postvaricella angiopathy (PVA), which demonstrates the imaging features of TCA but occurs within 12 months of clinical varicella infection [Askalan et al., 2001; Lanthier et al., 2005]). PVA also typically affects lenticulostriate arteries, with an irregular, unilateral stenosis centered on the proximal ACA or MCA that resolves over months [Askalan et al., 2001; Lanthier et al., 2005]. An example of this disease is shown in Figure 85-5. PVA is well established as occurring in adults following facial “shingles” [Gilden et al., 2009; Amlie-Lefond et al., 1995]. Proposed mechanisms include viral reactivation from the trigeminal ganglion, with spread along the trigeminovascular bundle that innervates proximal cerebral arteries [Kleinschmidt-DeMasters and Gilden, 2001]. Elevated cerebrospinal fluid antibody titers to varicella zoster virus are found rarely [Ueno et al., 2002], though cerebrospinal fluid polymerase chain reaction and/or antibodies may increase sensitivity [Kronenberg et al., 2002; Gilden et al., 2009]. Histopathological studies can reveal viral particles or antigens in cerebral artery smooth-muscle cells [Eidelberg et al., 1986; Linnemann and Alvira, 1980; Reshef et al., 1985], as was the case in two 4-year-old children who died of PVA-associated stroke [Berger et al., 2000]. With varicella vaccination becoming routine, naturally occurring PVA is expected to decrease. However, AIS has been described in proximity to varicella immunization [Wirrell et al., 2004].
The role of other infections in arteriopathic childhood AIS is undergoing active investigation. Fever and recent “viral infection” symptoms commonly are present in children with AIS. One study of childhood AIS reported fever in 50 percent and leucocytosis in 26 percent [Ganesan et al., 2003]. In another recent international study of 673 children with AIS, an association between viral upper respiratory tract infection and arteriopathy was found, particularly in children with TCA/FCA [Amlie-Lefond et al., 2009a]. Whether a common inflammatory pathophysiology underlies arteriopathy provoked by active viral infection, postviral infection, or “idiopathic” inflammation remains to be established definitively.
More obvious and malignant infections also predispose to ischemic stroke. Arterial or venous stroke associated with bacterial meningitis has been identified in 5–12 percent of children and as many as 75 percent of infants [Chiu et al., 1995; Silverstein and Brunberg, 1995; Chang et al., 2003]. Tuberculous meningitis is an important infectious cause of childhood AIS in certain countries [Springer et al., 2009]. Extension of subarachnoid meningeal infection and inflammation, causing thrombophlebitis of the perforating arteries, is the likely mechanism. Children with human immunodeficiency virus (HIV) often develop a unique arteriopathy that predisposes to both ischemic and hemorrhagic stroke [Patsalides et al., 2002; Visudtibhan et al., 1999; Kleinschmidt-DeMasters and Gilden, 2001]. While limited to small numbers of cases, AIS has been associated with mycoplasma, toxoplasmosis, Rocky Mountain spotted fever, Lyme disease, cryptococcosis, Japanese encephalitis, coxsackie B4 and A9, influenza A, enterovirus, parvovirus B19, and chlamydia [Guidi et al., 2003; Takeoka and Takahashi, 2002]. Regional infectious agents also must be considered, such as malaria, which was commonly associated with stroke in children in Cameroon [Obama et al., 1994]. Only a small portion of these studies have described the possible mechanism, such as TCA with enterovirus or HIV [Ribai et al., 2003; Leeuwis et al., 2007].
Arterial inflammation without a clear connection to active or previous infection is also a well-established form of cerebral arteriopathy, termed vasculitis. Recent reviews of childhood cerebral vasculitis are available [Elbers and Benseler, 2008]. Primary cerebral vasculitis is increasingly well described [Lanthier et al., 2001], and typically is referred to now as cPACNS [Elbers and Benseler, 2008; Benseler et al., 2006]. In contrast to “nonprogressive” cPACNS, which is very similar to TCA [Benseler et al., 2003; Elbers and Benseler, 2008], “progressive” cPACNS is frequently bilateral, and involves both proximal and distal cerebral arteries. Aggressive treatment may be required to prevent rapid deterioration and multifocal strokes. An example is shown in Figure 85-7. While the anterior circulation is involved more commonly, cPACNS diseases also can be isolated to the posterior circulation (Figure 85-8).
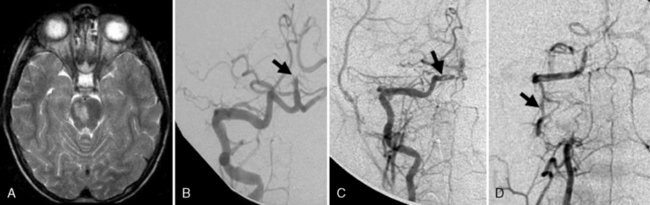
Some forms of cPACNS appear to be limited to distal, small cerebral arteries. Such small-vessel cPACNS lacks the acute stroke presentations described above, typically presenting with insidious symptoms such as headaches, cognitive decline, and personality change, and with MRI changes of multifocal enhancing lesions [Elbers and Benseler, 2008; Rafay et al., 2005; Benseler et al., 2005, 2006]. While conventional angiography is indicated, it may be negative in small-vessel cPACNS and brain biopsy is required for definitive diagnosis [Aviv et al., 2006; Benseler et al., 2005]. Small-vessel vasculitis may also be secondary to a variety of conditions, including infections, collagen vascular diseases, systemic vasculitides, and malignancies [Benseler and Schneider, 2004]. Systemic vasculitides and connective tissue diseases possibly associated with cerebral artery involvement include Kawasaki’s disease, Henoch–Schönlein purpura, polyarteritis nodosa, Wegener’s granulomatosis, systemic lupus erythematosus, Behçet’s syndrome, mixed connective tissue disease, Sjögren’s syndrome, and inflammatory bowel disease [Morfin-Maciel et al., 2002; Belostotsky et al., 2002; Berman et al., 1990; Morales et al., 1991; von Scheven et al., 1998; Graf et al., 1993; Benseler and Schneider, 2004]. Although systemic lupus erythematosus is often associated with AIS, vasculitis as the etiology for stroke is uncommon [Devinsky et al., 1988]. Large arteries can also be affected by secondary vasculitis, as in polyarteritis nodosa, or in Takayasu’s arteritis, where major branches off the aorta, including cervical arteries, are stenosed at their origin. Takayasu’s arteritis presents with asymmetrical and diminished pulses, renovascular hypertension, and stroke [Morales et al., 1991; Ringleb et al., 2005].
The range of infectious/inflammatory arteriopathies must be a primary consideration in all children with AIS, since they have important implications for investigation, treatment, and outcome. Diagnostic investigations should be tailored clinically, but may include screening for specific infections (serology, virology, polymerase chain reaction), in addition to routine hematological blood work, and inflammatory (erythrocyte sedimentation rate, C-reactive protein) and vasculitic (antinuclear antibodies, extractable nuclear antigens, complement, etc.) markers. In primary cPACNS, results of blood and cerebrospinal fluid studies are often negative [Elbers and Benseler, 2008; Riou et al., 2008]. More advanced studies to determine the markers and the relationship between infarction, inflammation, and arteriopathic AIS in children are under way. No specific treatment trials have been performed for the infectious/inflammatory cerebral arteriopathies in children. Whether an antiviral agent like acyclovir could alter the clinical course of PVA has not been determined but should certainly be considered in an immunocompromised child. Once a noninfectious inflammatory vasculitis is diagnosed, immunosuppression, with agents including corticosteroids, cyclophosphamide, azathioprine, or others, should be considered [Benseler and Schneider, 2004; Elbers and Benseler, 2008]. The advice of a pediatric rheumatologist in diagnosing and treating inflammatory cerebral arteriopathy should be sought.
Dissection and other physical injury
Craniocervical dissection represents a breach (usually mechanical) in arterial wall integrity with formation of a subintimal hematoma. AIS complicates arterial dissection by several mechanisms, including arterial occlusion at the dissection site by intraluminal thrombosis or wall hematoma, distal extension of the dissection along the artery, or, most frequently, by artery-to-artery thromboembolism. Dissection in the carotid or posterior vertebrobasilar circulation accounts for up to 20 percent of childhood AIS [Chabrier et al., 2003; Fullerton et al., 2001; Camacho et al., 2001; Rafay et al., 2006]. Extracranial arterial dissection occurs at predictable locations, including the proximal internal carotid artery and in the vertebral arteries at the C1–C2 level. Intracranial dissection can also occur, and carries the added risk of secondary subarachnoid hemorrhage [Fullerton et al., 2001]. Single-center series applying strict diagnostic criteria for dissection report that extracranial dissection is three times more frequent than intracranial dissection in children [Rafay et al., 2006]. A pooled analysis of published cases reported intracranial dissection to be more common [Fullerton et al., 2001], though distinguishing this from TCA is often impossible.
Dissection may result from trauma to the neck, spine, or retropharyngeal area secondary to obvious mechanical injuries or more subtle insults, including external manipulation, physical exertion, or contact sports [Rafay et al., 2006; Chabrier et al., 2003]. However, many appear to occur “spontaneously,” and with minor trauma being so common in children, a definitive causative event is often lacking. Adult studies have suggested that inherent differences in connective tissue structure, detectable on skin biopsy (but not physical examination) may predispose to arterial dissection [Martin et al., 2006]. Nonaccidental trauma should also be considered in small children with dissection [Agner and Weig, 2005]. New-onset head or neck pain is a prominent presenting feature in children, but clinical stroke symptoms may have their onset 7–10 days following pain or the traumatic event. Additional clinical findings may include an ipsilateral Horner’s syndrome, cervical bruit, or musculoskeletal cervical spine abnormalities. Recurrence rates are substantial in dissection, estimated at 20 percent or higher [Rafay et al., 2006; Fullerton et al., 2001, 2007b; Chabrier et al., 2003].
Whether dissection affects the intracranial or extracranial vessels, the medical management of dissections is controversial. Based on theoretical reasons, consideration of an anticoagulation approach is suggested by consensus guidelines [Monagle et al., 2008b; Roach et al., 2008]. Intracranial dissection is considered a relative contraindication due to the risk of subarachnoid hemorrhage. Adult dissection traditionally has been treated with anticoagulation; however, no randomized trials have been conducted and antiplatelet strategies have been suggested recently [Engelter et al., 2007]. Retrospective studies suggest that both are likely safe with generally good outcomes [Engelter et al., 2000]. An adult randomized trial appears logistically unfeasible, suggesting that a pediatric version will never occur. Long-term complications of pediatric dissection, such as recurrent stroke or pseudoaneurysm, require surveillance and further study [Tan et al., 2009a]. Activity restrictions and return-to-play guidelines following dissection are lacking, though pediatric stroke experts appear reluctant to recommend return to high-impact activities [Bernard et al., 2007]. A graded return to activity over months, with avoidance of high-risk activities (contact sports, chiropractic manipulation), is suggested.
Additional acquired vascular injuries may cause childhood AIS. Postradiation vasculopathy manifests with progressive large-vessel stenosis and cerebral ischemia years after irradiation. Younger children with optic chiasm gliomas or other sellar region tumors exposed to higher-dose, focused radiation are at increased risk [Omura et al., 1997]. MRI may demonstrate arterial wall thickening and contrast enhancement [Aoki et al., 2002]. Young adult survivors of childhood cancers recently were shown to harbor a higher risk of stroke [Jordan and Duffner, 2009a]. AIS also may occur in the setting of rapid brain expansion due to diffuse edema, space-occupying lesion, or hydrocephalus, where mechanical compression and occlusion of the anterior (subfalcine) or posterior (tentorial edge) cerebral arteries may occur. Stroke associated with catheterization of cerebral vessels for extracorporeal membrane oxygenation (ECMO) also has been described, with evidence of AIS in 50 percent of 44 autopsied children dying after ECMO [Jarjour and Ahdab-Barmada, 1994].
Moyamoya disease and syndrome
Moyamoya describes the progressive occlusion of the cerebral arteries at the circle of Willis, leading to formation of abnormal networks of small collateral vessels with a “puff of smoke” appearance on angiography (Figure 85-9) [Scott and Smith, 2009; Kuroda and Houkin, 2008]. The classic pattern involves the distal internal carotid arteries bilaterally, but unilateral variants and involvement of additional anterior and posterior circulation vessels are well described. Moyamoya disease is the idiopathic version described primarily in Asian populations, while moyamoya syndrome occurs in association with other conditions, including sickle cell disease (see below), Down syndrome, neurofibromatosis type 1, Williams’ syndrome, postradiation vasculopathy, and congenital arteriopathies (see below) [Jea et al., 2005; Scott and Smith, 2009]. Familial forms of moyamoya increasingly are described, with several potential genetic mechanisms proposed [Ikeda and Yoshimoto, 2005]. Moyamoya is, therefore, a heterogeneous syndrome and not a single disease, but recognizing the shared patterns of presentation is clinically relevant to ensure accurate investigation and treatment.
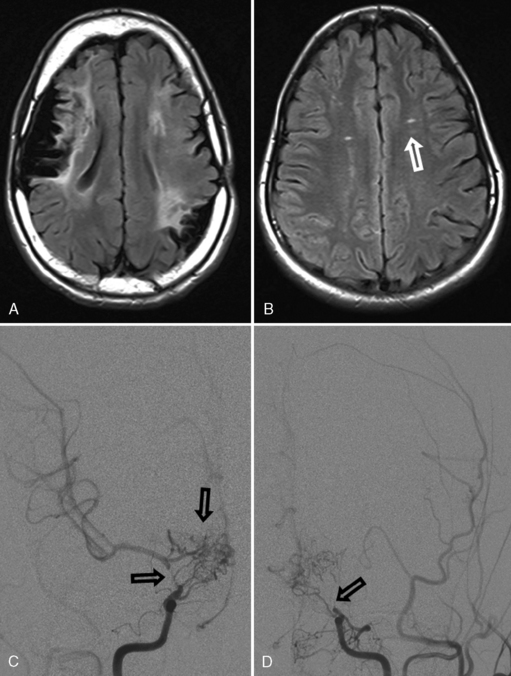
Moyamoya presents with unique clinical syndromes and patterns of stroke. In contrast to most other forms of AIS, an important mechanism for moyamoya-associated infarction is hypoperfusion. In the setting of internal carotid artery occlusion, perfusion is compromised most severely in the distal perfusion zones of the MCA and ACA in the deep hemispheric white matter. As a result, small (<1 cm) collections of adjacent infarcts accumulate in these “deep watershed” zones. Impaired cerebrovascular autoregulation in chronically underperfused brain territories appears to be an important underlying mechanism for these infarcts [Mikulis et al., 2005]. Therefore, TIA symptoms may be precipitated by alterations in PCO2, such as during hyperventilation (cerebral vasoconstriction) or breath holding (cerebral vasodilatation with “steal”). These infarcts may be subclinical, often with gradual cognitive or behavioral decline, although clear stepwise neurological deteriorations may occur. Moyamoya progresses slowly over time, causing accumulating injury and poor neurological outcomes [Suzuki and Kodama, 1983]. Progression of moyamoya may be faster in preschool-aged children [Kim et al., 2004]. In addition, artery-to-artery embolism of larger cerebral arterial branches can result in more typical AIS in moyamoya. Hemorrhagic stroke also can occur from the compensatory basal collateral vessels (“rete mirabile”), and this risk increases in early adulthood [Yoshida et al., 1993]. Headaches, often with migrainous features, are common in moyamoya and may precede other neurological symptoms.
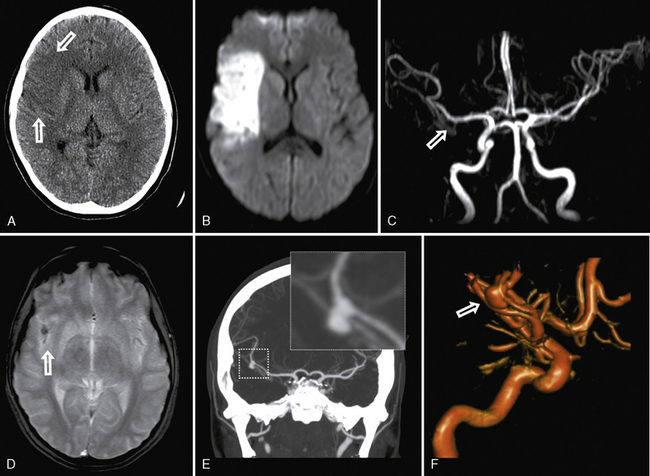
MRI can suggest moyamoya with small flow voids in the basal ganglia and the infarct patterns described above. MR angiography is highly but incompletely sensitive at detecting moyamoya arteriopathy. Advanced fMRI and other perfusion studies may demonstrate impaired autoregulation [Mikulis et al., 2005; Mori et al., 2009]. However, conventional angiography is required in all children with moyamoya to confirm the presence of collaterals, characterize disease extent and severity, and image the external carotid circulation to determine options for revascularization surgery (see Figure 85-9) [Satoh et al., 1988]. Hypertension is common in moyamoya and may be secondary to associated renal artery stenosis [Kirton et al., 2006]. Doppler ultrasound and/or renal angiography during cerebral studies are indicated in such patients to exclude renovascular involvement.
Treatment of moyamoya is complex. Antiplatelet therapy with acetylsalicylic acid (ASA) often is employed and is likely safe. The chronic, progressive occlusion of distal internal carotid arteries, and consequent low perfusion, result in cerebral autoregulation shifting toward maximal vasodilatation in order to maintain cerebral perfusion. Carbonic anhydrase inhibitors, including acetazolamide, may shift acid–base balance to favor vasodilatation in moyamoya [Ikezaki, 2000]. However, in severe moyamoya, such medications may cause a “steal” or “reverse Robin Hood” phenomenon, whereby dilatation of healthier arteries diverts perfusion away from the most critically affected areas, which subsequently infarct. Such interventions must be considered carefully in severely affected children. Use of antihypertensives in children with moyamoya and hypertension must also be considered carefully, as even small decreases in cerebral perfusion can lead to infarction.
Surgical revascularization procedures are highly successful at improving cerebral perfusion in children with moyamoya [Scott and Smith, 2009]. Direct procedures connect extracranial blood vessels, such as the superficial temporal artery, to distal portions of intracranial vessels, usually the MCA. Direct revascularizations are technically limited in small children and carry some risk of reocclusion. In children, indirect procedures are now employed more frequently where extracranial vessels are approximated to the cortical surface overlying areas of decreased perfusion. Angiogenic signals from ischemic parenchyma subsequently result in neovascularization of the affected areas over a course of months. Encephalodural synangiosis (EDAS) and encephalodural myosynangiosis (EDMS) are common procedures transferring extracranial arteries alone or within scalp muscle, respectively, to the pial surface. A study of 143 children undergoing pial synangiosis found that more than 90 percent enjoyed symptom-free survivals with follow-up of more than 5 years [Scott et al., 2004]. Studies comparing the different surgical approaches are lacking. Indirect procedures are now used widely and sometimes combined with direct methods [Fung et al., 2005]. Perioperative infarctions likely occur in less than 10 percent of cases [Fung et al., 2005; Kim et al., 2004]. Younger age of onset in moyamoya may carry an increased risk of recurrent stroke and poor outcome, suggesting that early surgery is indicated [Kim et al., 2004]. Preliminary evidence suggests revascularization also may be helpful in some cases of sickle-related moyamoya [Hankinson et al., 2008]. Success of revascularization surgery can be evaluated with serial angiography, as well as new cerebrovascular reserve MRI methods [Mikulis et al., 2005].
Cardiac Risk Factors
Heart disease is a major risk factor for AIS in infancy and childhood, present in 12–28 percent of cases [Chabrier et al., 2000; Ganesan et al., 2003; deVeber, 2000]. Abnormalities in cardiac structure, flow, rhythm, endothelium, ventricular function, valves, blood viscosity, and other factors may contribute to thrombus generation and subsequent cerebral embolization. Multiple AIS in multiple, distinct vascular territories (e.g., left and right) suggests a cardiac source. Complete cardiac evaluations should be expedited in acute stroke of unknown etiology, to guide acute medical management and emergency cardiac treatment when indicated.
Complex congenital heart disease, particularly cyanotic lesions, carries the highest stroke risk. Both catheterization procedures and surgical repairs increase risk further. A recent single-center case-control study of more than 5500 children undergoing cardiac surgery documented a symptomatic AIS risk of 5.4 per 1000 (1:185), with reoperation being an independent risk factor [Domi et al., 2008]. This risk is comparable to or higher than the figure in previous studies [Menache et al., 2002]. Certain surgeries carry elevated short- and long-term risk. With the Fontan procedure, risk estimates of immediate or delayed stroke range from 3 to 19 percent [Monagle and Karl, 2002; Barker et al., 2005; Kaulitz et al., 2005]. Risk of symptomatic stroke following cardiac catheterization in children is estimated at 1:600–700 [Liu et al., 2001] with interventional more risky than diagnostic catheterizations [Hamon et al., 2006]. Balloon atrial septostomy prior to transposition switch surgery carries a stroke risk approaching 50 percent, according to one study of 29 patients [McQuillen et al., 2006]. Many ischemic brain lesions related to congenital heart disease are asymptomatic in young children and often are present preoperatively [Miller et al., 2004].
Intracardiac septal defects, such as atrial septal defect and patent foramen ovale, remain controversial risk factors for stroke in the young [Kizer and Devereux, 2005] and have not been well studied in children. Atrial septal lesions create the potential for intermittent right-to-left intracardiac shunting with paradoxical embolization of systemic venous clots to the brain [DiTullio et al., 1992]. When such right-to-left intracardiac shunts are detected, further investigation for systemic venous thrombosis may be indicated [Mas et al., 2001]. Septal lesions such as patent foramen ovale are common in the pediatric population. Associations between patent foramen ovale and otherwise unexplained stroke have been demonstrated repeatedly in young adults [Mas et al., 2001; Handke et al., 2007], although patent foramen ovale may not increase the risk of recurrent stroke [Almekhlafi et al., 2009]. A recent adult analysis suggests that a high proportion of patent foramen ovale in young cryptogenic stroke patients are likely incidental [sheikh-Ali et al., 2009]. Patent foramen ovale associated with atrial septal aneurysm [Mas et al., 2001] and thrombophilias carries increased stroke risk [Pezzini et al., 2003; Rodriguez and Homma, 2003]. The indications for medical and surgical (closure) treatment of uncomplicated patent foramen ovale remain controversial [Tamayo and Harrer, 2009].
Several acquired cardiac etiologies predispose to AIS. Infective endocarditis carries a high risk of multifocal embolic AIS and risk of mycotic aneurysm and intracerebral hemorrhage (Figure 85-10). Detection of signs of infective endocarditis, including fever, physical markers (Janeway lesions, Roth spots), and history of subacute constitutional symptoms or recent dental work, enables early diagnosis and treatment. Anticoagulation is relatively contraindicated in infective endocarditis due to bleed risk from mycotic aneurysms, and recent adult studies have demonstrated frequent subclinical microbleeds with susceptibility-weighted MRI [Klein et al., 2009]. Other acquired cardiac etiologies include poor left ventricular function secondary to cardiomyopathy (infectious, metabolic) [Hays et al., 2006], valvular heart disease (e.g., rheumatic fever, artificial valves), arrhythmia, and atrial myxoma [Omeroglu et al., 2006].
All children with AIS of undetermined cause should have prompt cardiac evaluation, including electrocardiogram and echocardiography. Agitated saline “bubble studies” can help establish the presence of patent foramen ovale and atrial septal defects. In adults, transesophageal echocardiography is superior to transthoracic echocardiography in diagnosing a cardiac stroke source, detecting 31 percent more treatable lesions [de Abreu et al., 2008; de Bruijn et al., 2006]. However in young children with a thin chest wall, transthoracic echocardiography may suffice. The roles of cardiac MRI and ambulatory cardiac rhythm monitoring in childhood AIS have not been investigated.
Consensus across published guidelines suggests that proven or suspected cardiogenic AIS requires specific management [deVeber and Kirkham, 2008]. With a presumed dominant role of the coagulation system, acute anticoagulation with low-molecular-weight heparin (LMWH) usually is indicated. If detected, bacterial endocarditis also requires urgent antibiotic treatment and investigation for mycotic aneurysm [Venkatesan and Wainwright, 2008]. Duration of anticoagulation depends on individual factors that determine recurrence risk, including circumstances of the initial stroke (spontaneous or procedure-related), corrected or uncorrected cardiac defect, prothrombotic testing results, and others.
Prothrombotic and Hematological Disorders
Thrombophilia, or prothrombotic disorders, refers to abnormalities of the coagulation, fibrinolytic, and platelet systems that predispose to pathological thrombus formation. Studies in childhood stroke are complicated by the developmental evolution of the hemostatic system in childhood, a complex variety of disorders, few case-control studies, and inconsistent testing and laboratory methods. With these limitations in mind, a role for prothrombotic disorders likely exists in childhood AIS, though most patients likely have an additional “triggering” risk factor at the time of AIS [Barreirinho et al., 2003; Strater et al., 2002]. Detailed reviews are available [Mackay and Monagle, 2008; Barnes and deVeber, 2006].
Inherited or acquired coagulation disorders have been identified in 20–50 percent of children with AIS [Bonduel et al., 1999; deVeber et al., 1998b; Barnes and deVeber, 2006; Herak et al., 2009]. Factor V Leiden is one of the strongest associations [Strater et al., 2002], with a meta-analysis of five case-control studies suggesting a combined odds ratio of 4.3 for stroke with heterozygote factor V Leiden [Chan et al., 2000]. Elevated levels of lipoprotein (a) and Protein C deficiency are both more common in children with AIS compared to controls [Nowak-Goettl et al; 1999; Barnes and deVeber, 2006]. Although mutations in methylene tetrahydrofolate reductase (MTHFR) are associated with childhood stroke, they are also common in the population and their significance without hyperhomocysteinemia is unclear [Nowak-Goettl, 1999; Ganesan et al., 2003; Mackay and Monagle, 2008]. In contrast to most prothrombotic states that promote venous thrombosis, prothrombin G20210A mutations are associated more frequently with arterial thrombosis in children [Young et al., 2003]. Meta-analysis of studies to date suggests an odds ratio of 3.5 in childhood AIS [Barnes and deVeber, 2006]. Case-control studies also have confirmed an increased occurrence of antiphospholipid antibodies and lupus anticoagulant in childhood AIS [Kenet et al., 2000; Ganesan et al., 2003].
A systematic review of published case-control data has reported increased pooled odds ratios for childhood AIS for protein C deficiency (6.5) and methylene tetrahydrofolate reductase (MTHFR C677T) (1.7), but ratios were not significant for protein S deficiency (1.14), AT (antithrombosis) deficiency (1.02), APCr (activated Protein C resistance) (1.34), prothrombin G20210A (1.10), or elevated plasma homocysteine (1.36) [Haywood et al., 2005]. Presence of certain prothrombotic states increases stroke recurrence risk in children, including factor V Leiden, protein C deficiency, and elevated lipoprotein (a) [Ganesan et al., 2006; Strater et al., 2002], but not elevated ACLA (anticardiolipin antibody) antibodies [Lanthier et al., 2004]. Plasminogen activator inhibitor abnormalities do not appear increased in childhood AIS [Nowak-Gottl et al., 2001]. Additional prothrombotic factors of interest include abnormal levels of plasminogen, fibrinogen, and coagulation factors (VIII, IX, XI), and an increasing number of genetic polymorphisms. Although platelet abnormalities are likely of importance in arterial thrombosis, only limited studies have been conducted in childhood AIS [Herak et al., 2009; Barnes and deVeber, 2006].
Sickle cell disease
Sickle cell disease (SCD) is the most common hematological disorder associated with cerebrovascular disease in children and increases ischemic stroke risk 400-fold. Overall, about 25 percent of SCD children develop cerebrovascular complications, including overt AIS, subclinical small AIS, moyamoya, or hemorrhagic stroke [Pegelow, 2001]. Strokes are ischemic in 75 percent and hemorrhagic in 25 percent, the latter increasing with age [Satoh et al., 1988]. Stroke is responsible for 10 percent of SCD mortality [Manci et al., 2003]. There are two primary mechanisms for AIS in SCD. First, a progressive internal carotid (large-vessel) moyamoya arteriopathy can develop with typical moyamoya-type deep watershed or large-artery territory infarcts (see moyamoya above). The etiology of this arteriopathy is thought to relate to the chronic effects of increased carotid blood-flow rates with sickled red cells damaging endovascular surfaces. Recent studies indicate that proinflammatory genetic polymorphisms also may play a role [Hoppe et al., 2007]. Second, SCD may be associated with occlusion of small cerebral end-arteries by sickled cells and related thrombosis, resulting in randomly distributed multifocal small infarcts. Stroke may occur at any time in SCD, though risk is highest during an acute crisis.
Transcranial Doppler studies are a noninvasive means of following flow rates related to large-vessel vasculopathy, with flows above 200 cm/s predicting increased stroke risk [Adams et al., 1992, 1998]. The STOP (Sickle Cell Transfusion Stroke Prevention) randomized controlled trial demonstrated that regular transfusion therapy (hemoglobin S below 20–30 percent) produced a 90 percent relative risk reduction in initial stroke in children with elevated velocities [Adams et al., 1998]. Recurrent stroke rates were also reduced. A follow-up study showed that stroke risk returns with discontinuation of transfusions [Adams et al., 2005]. This suggests that most families currently need to continue with arduous blood transfusions every 3–6 weeks, including chelation therapy and screening for iron overload [Monagle et al., 2008b; Roach et al., 2008; Aygun et al., 2009]. A similar trial looking at children with small vessel SCD is under way (Silent Infarct Transfusion [SIT] trial; http://sitstudy.wustl.edu/). Increased ultrasound screening and decreased stroke rates have been documented since STOP was published [Fullerton et al., 2004; Armstrong-Wells et al., 2009].
In addition to annual TCD (Trancranial Doppler) screening beginning at age 3, serial MRI should be considered, as this can detect silent infarcts secondary to small-vessel disease. MR angiography should be considered following large-vessel disease and possible moyamoya [Zimmerman, 2005]. Additional stroke prevention strategies in SCD currently under evaluation include hydroxyurea [Heeney and Ware, 2008; Ware et al., 2004] and nocturnal oxygen supplementation [Kirkham et al., 2001b]. Antithrombotic therapy has not been studied but aspirin should be considered, especially in cases with large-vessel vasculopathy, prothrombotic abnormalities, or recurrent stroke [Wolters et al., 1995]. Revascularization surgery may be successful in SCD-related moyamoya [Fryer et al., 2003; Hankinson et al., 2008]. Bone marrow transplantation is feasible in SCD [Walters et al., 1996], but may carry an increased risk of neurological complications in children with cerebrovascular disease [Walters et al., 1995].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


