Heredodegenerative Diseases
John H. Menkes
Advances in molecular biology have pointed to a basic similarity between diseases of metabolic origin (see Chapter 1) and heredodegenerative diseases. Nevertheless, essential differences remain. In metabolic disorders, the biochemical derangement of tissue or body fluids has led, in enzymopathies such as phenylketonuria or maple syrup urine disease, to the identification of the basic biochemical defect. Subsequent purification of the normal protein has permitted isolation of the gene that codes for it and has led to the determination of the molecular abnormality or abnormalities in the gene in patients with the disease. This process has been termed functional cloning.
Many of the conditions covered in this chapter do not yet have a biochemical explanation. This lack has prompted the application of positional cloning (reverse genetics, a process in which the gene responsible for the disorder is isolated without its protein product being known). Theoretically, the process is simple; the approximate map position of the gene is established by linkage studies on extended families experiencing the disease. In many instances, cytogenetics and the use of a large number of polymorphic markers allow assignment of the gene to a specific chromosome with a resolution of roughly several million base pairs. This first step has been accomplished for quite a few of the conditions covered in this chapter. The second step involves a more exact localization of the gene site and subsequent isolation and cloning of the gene. Although this has been a complicated and labor-intensive process, it has been achieved for a significant proportion of the heredodegenerative diseases covered in this chapter.
A shortcut to this process, which is also applicable to the rarer diseases, has been the use of candidate genes. A number of genes coding for proteins and enzymes vital to brain function have been mapped, cloned, and sequenced. When a new disease locus is assigned to a chromosomal region using linkage analysis, candidate genes from the same region are surveyed and features of these candidate genes are compared with the features of the particular disease. Conversely, when a new gene is assigned to a chromosomal region, candidate diseases are analyzed by an analogous process. This approach has led to the observation that abnormalities in the gene for myelin proteolipid protein (PLP) are responsible for Pelizaeus-Merzbacher disease (PMD), with the gene for both PLP and the disease localized to the q22 segment of the X chromosome. One of the genes for Charcot-Marie-Tooth (CMT) disease [hereditary motor sensory neuropathy (HMSN) type IA] was isolated by this combined approach.
The interested reader is referred to reviews of this constantly evolving field in Chapters 14 (Identifying Human Disease Genes) and 16 (Molecular Pathology) in the text by Strachan and Reed (1) and Chapter 1 (Introduction to Medical Genetics and Methods of DNA Testing) in the book edited by Pulst (2).
For the clinician, it is still best that the heredodegenerative diseases are grouped according to their clinical presentation and according to the site in the brain that incurs the major anatomic or functional insult. The categories are (a) heredodegenerative diseases of the basal ganglia, (b) heredodegenerative diseases of the cerebellum, brainstem, and spinal cord, (c) heredodegenerative diseases of the peripheral and cranial nerves, and (d) diffuse cerebral degenerative diseases. A variety of intermediate forms have been well documented.
As our knowledge of the interrelation between genotype and phenotype expands it has become evident that many common neurologic phenotypes have multiple genetic etiologies and, conversely, that there are many examples of mutations in single genes that produce distinct phenotypes. This chapter will provide examples of both phenomena. Thus there are at least 21 genetic forms of autosomal dominant cerebellar ataxias, and the Charcot-Marie-Tooth phenotype results from mutations in more than a dozen different genes. On the other hand, different mutations in the voltage-dependent calcium-channel gene (CACNA1A) can produce one of the dominant spinocerebellar ataxias (SCA6), episodic ataxia (EA2), or familial hemiplegic migraine. Even when there appears to be a clear relationship between a mutated gene and its phenotype, the correlation between genotype and phenotype is far from complete, and there are often known or unknown genetic or environmental phenotypic modifiers that affect
penetrance and expression. One example of this perplexing situation is the situation in DYT1 dystonia, one of the primary torsion dystonias, in which the question of why dystonia is only present in some 30% of gene carriers is completely unresolved. Another such example is the recently uncovered effect of genetic modifiers on the splicing machinery that can result in changes in the levels of normal transcripts or in the relative ratios of wild type to mutant transcripts such as has been documented for familial dysautonomia.
penetrance and expression. One example of this perplexing situation is the situation in DYT1 dystonia, one of the primary torsion dystonias, in which the question of why dystonia is only present in some 30% of gene carriers is completely unresolved. Another such example is the recently uncovered effect of genetic modifiers on the splicing machinery that can result in changes in the levels of normal transcripts or in the relative ratios of wild type to mutant transcripts such as has been documented for familial dysautonomia.
HEREDODEGENERATIVE DISEASES OF THE BASAL GANGLIA
Huntington Disease
Huntington disease (HD) is a chronic progressive degenerative disease characterized by involuntary movements, mental deterioration, and an autosomal dominant transmission. Although its essential features were first described by Waters (3) in 1841, Huntington provided its most accurate and graphic description in residents of East Hampton, Long Island (4).
The prevalence of HD in the general population has been estimated at between 5 and 10 per 100,000 (5). Although all patients early in the history of the United States were descendants of two English brothers, the disease has since been found in all races and nationalities.
Morphologic abnormalities are limited to the brain. Grossly, patients have gyral atrophy affecting primarily the frontal lobes and generalized and symmetric dilatation of the lateral ventricles. The striatum (caudate nucleus and putamen) is markedly shrunken, with the tail of the caudate nucleus being the area that is most affected (Fig. 3.1). Light microscopic examination demonstrates three processes: neuronal loss, which is associated with the presence of brain macrophages and neuronal satellitosis; lipopigment deposition in neurons and astroglia in excess of that expected for the patient’s age; and a relative astrocytic proliferation (6). In adults, these changes are found in the caudate nucleus, putamen, and, to a lesser extent, the cortex, notably the frontal lobes (7). The ventrolateral thalamic nucleus, subthalamic nucleus, and, in some cases, spinal cord also may be involved. As a rule, the caudate nucleus, particularly its periventricular portion, is more affected than the putamen, whereas the nucleus accumbens generally is spared until late in the disease process.
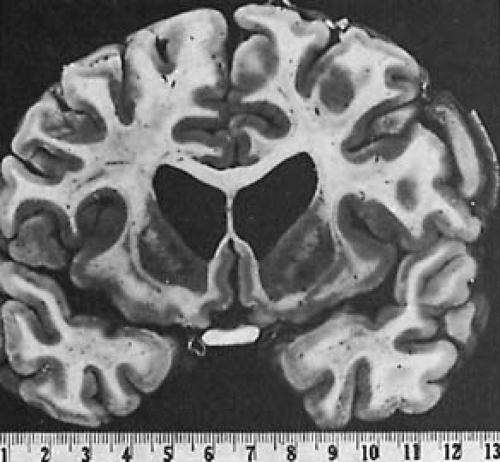 FIGURE 3.1. Huntington disease. Gross atrophy of the caudate nucleus gives the lateral ventricles a butterfly appearance. (Courtesy of Dr. J. A. N. Corsellis, The Maudsley Hospital, London.) |
The pathology of juvenile HD is somewhat similar. Children presenting with the rigid form of the disease appear to have a marked destruction of the putamen (8,9). Severe astrogliosis of the globus pallidus may occur without neuronal loss, and hyperpigmentation of the substantia nigra has been noted in some juvenile patients (10).
Electron microscopy of frontal lobe biopsies reveals a generalized, nonspecific derangement of all membrane structures, a consequence of cell death.
Cell death within the striatum does not affect all neurons equally. The medium-sized gamma-aminobutyric acid (GABA)-ergic spiny projection neurons, which make up approximately 95% of human striatal neurons and contain a variety of neuropeptides, notably enkephalins and substance P, are the first to be affected (11). They are the source of the striatal output projections to the external globus pallidus and to the substantia nigra pars reticulata. By contrast, medium-sized aspiny interneurons containing somatostatin and staining with the histochemical marker nicotinamide adenine dinucleotide phosphate (NADPH)–diaphorase are relatively spared, as are the large, aspiny, cholinergic neurons and neurons immunoreactive for neuropeptide Y.
Selective cell loss also is observed in the cortex, where a depletion of the large pyramidal neurons in cortical layers III, V, and VI and sparing of the GABA-ergic local circuit neurons occur (12). During the initial phases of the disease, cell loss is at least partially compensated by augmentation of the dendritic trees of the surviving pyramidal cells. This observation could explain the normal cortical metabolic activity that is observed in the early phase of the illness (13).
Pathogenesis and Molecular Genetics
The gene for HD has been mapped to the short arm of chromosome 4 (4p16.3) (14). This work represented the first successful linkage analysis in humans using polymorphic DNA markers. The gene whose mutation causes HD is widely expressed in all regions of normal and HD brain and in various other organs. It encodes a large cytoplasmic protein named huntingtin (15). Wild-type huntingtin is essential for normal embryogenesis and the formation of forebrain (16).
HD is an autosomal dominant condition; therefore one mutated gene suffices to cause the disease. Because hemizygous loss of one of the two Huntington genes—as occurs in Wolf-Hirschhorn syndrome—does not produce an HD phenotype, it is evident that the mutation confers a deleterious function on the mutant huntingtin. The disease is now known to result from an expansion of a trinucleotide repeat in the coding region of the gene (17). Trinucleotide repeats are repeated units of three nucleotides in genomic DNA. In the case of HD, it is the trinucleotide (triplet) cytosine-adenine-guanine (CAG) that is repeated. Whereas in the normal population this trinucleotide repeat ranges from 9 to 34 copies, the length of this repeat is expanded in HD to a mean of 42 to 46 copies. This long run of CAGs in the HD gene encodes a polyglutamine tract near the amino terminus of huntingtin. In HD the length of the repeat is highly unstable. Whereas during maternal transmission it changes by only a few repeats and on rare occasions has been shown to contract, in paternal transmission the length of the repeat increases dramatically in approximately one-third of cases. Of patients with repeats of 55 or more, 90% have an onset of the disease by age 30 years, and a good negative correlation exists between the length of the repeat and the age of onset of neurologic symptoms, with the longest repeats resulting in juvenile HD (18).
When HD appears sporadically, it is often the result of an expansion from an already large repeat. Such an expansion is more likely to occur on the paternally derived allele and has been correlated with advanced paternal age (19). The basic processes responsible for the instability of some repeat trinucleotide DNA sequences, a process termed dynamic mutation, are unclear (20).
A similar expansion of genes with triplet repeats is seen in a number of neurologic conditions (Table 3.1). It would now appear that whenever a dominantly transmitted heredodegenerative disease manifests genetic anticipation (i.e., whenever a progressively earlier onset of neurologic symptoms occurs in successive generations), a repeat expansion of trinucleotide repeats is suspected (21) (see Chapter 4, Fig. 4.5). However, Fraser stressed that ascertainment bias, that is, that the younger generation has not yet reached the age when the disease becomes manifest, and truncation bias, meaning that parents with an early onset of disease do not have offspring, also should be considered as a cause for anticipation in neurologic conditions (22). In addition, a high incidence of repeat expansions occurs in the normal population, and between 30% and 35% of humans have CAG repeat lengths in their genomes that exceed 40 repeats, an indication that trinucleotide expansion is not invariably associated with a disease process.
It is not fully understood how the expanded triplet repeat on the Huntington gene causes the disease. Several investigators have observed neuronal intranuclear inclusions immunoreactive for huntingtin epitopes in cortical and striatal neurons of HD brain (23). These inclusions comprise truncated derivatives of the mutated huntingtin and are also found in presymptomatic HD (24). It is likely that the expanded polyglutamine tract alters the physical properties of huntingtin in such a manner as to induce its accumulation within the cell nucleus. This contrasts with the cytoplasmic location of wild-type huntingtin (24a). Nuclear accumulation of mutant huntingtin induces cell dysfunction and ultimately neuronal death. Of note is the presence of ubiquitin in these inclusions. Ubiquitin is a small protein that is crucial for the degradation of many cytosolic nuclear and endoplasmic reticulum proteins (25). Because binding of ubiquitin to proteins marks them for subsequent degradation, this finding suggests that mutant huntingtin has been targeted for proteolysis but that the degradation was somehow incomplete.
TABLE 3.1 Triplet-Repeat Diseases | |||
|---|---|---|---|
|
How mutant huntingtin induces a selective cell death is the subject of much research. More than a dozen proteins interact with the amino terminal of huntingtin. Some are involved in intracellular signaling; others are involved in transcriptional events, and the function of yet others is poorly characterized (26,27). Experimental work suggests that polyglutamine-expanded huntingtin acts within the nucleus to induce apoptotic cell death (28). Antiapoptotic compounds protect neurons against the mutant huntingtin, as does blocking nuclear localization of mutant huntingtin (29).
The mechanisms for selective vulnerability that is so characteristic for HD are unclear. Current thinking is that overactivity of the N-methyl-D-aspartate (NMDA)-type glutamate receptors, particularly the NR2B subtype, is at least contributory in that it can result in excitotoxicity-induced
cell death in the medium-sized spiny GABA-ergic projection neurons that are enriched in the NR2B receptors (30). In support of this hypothesis is the marked reduction in NMDA-receptor binding in the putamen of brains in patients with HD as compared with normal brains (31).
cell death in the medium-sized spiny GABA-ergic projection neurons that are enriched in the NR2B receptors (30). In support of this hypothesis is the marked reduction in NMDA-receptor binding in the putamen of brains in patients with HD as compared with normal brains (31).
It is not clear whether mitochondrial dysfunction and consequent oxidative stress represent primary abnormalities or are secondary to early neuronal dysfunction (32). Respiratory complex II/III is deficient in putamen derived from patients with HD but is normal in their cerebral cortex, cerebellum, and cultured fibroblasts. This observation suggests that mitochondrial dysfunction parallels the severity of neuronal loss and could be secondary to cell damage induced by excitotoxicity and the ensuing generation of free radicals and superoxide (33).
Clinical Manifestations
Although symptoms usually begin between ages 35 and 40 years, approximately 5% of patients are younger than 14 years (34). In general, the age when symptoms have their onset is inversely related to the CAG repeat number (35). However, the age of onset is also a function of genes other than the HD gene and is partially environmentally determined (36). Age of onset also decreases as the number of affected male ancestors increases. This finding is most striking when both the affected parent and grandparent are men. In a large series of patients with juvenile HD (patients whose symptoms become apparent before 20 years of age), Myers and coworkers found that 83% inherited the disease from their fathers (34). The onset of neurologic symptoms in siblings of patients with juvenile HD is approximately 27 years. This timing is significantly earlier than in the general population of patients with HD (37). The best explanation for the phenomenon of paternal inheritance of juvenile HD is that an expansion of the triplet repeat occurs during spermatogenesis.
The clinical picture of HD as it presents in children differs from that of affected adults (Table 3.2).
TABLE 3.2 Clinical Features of Huntington Disease of Childhood (28 Cases) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
As surveyed by Cotton and colleagues (38), the initial stages of the illness are marked by mental deterioration or behavioral abnormalities (20 of 40 patients), gait disturbances, usually the consequence of rigidity (18 of 40 cases), cerebellar signs (15 of 40 cases), and, less commonly, seizures. In the Dutch series of Siesling and coworkers, behavioral disorders were the first feature of juvenile HD in 70% and chorea was seen in 48% (39). Some 70% of children in this series who presented with chorea developed rigidity in the course of the disease. Mental deterioration can commence before 2 years of age (40). It is not clear whether the intellectual decline results from striatal or cortical dysfunction.
The clinical onset of rigid cases is earlier than in patients who have chorea as the predominant movement disorder (39). Additionally, in almost 90% of rigid juvenile HD cases, the disease is paternally inherited. This contrasts with approximately 70% for juvenile HD with chorea (39,41).
When present, choreoathetosis resembles that seen in Sydenham chorea, but the movements have a greater affinity for the trunk and proximal musculature and, unlike those in Sydenham chorea, are rarely asymmetric. They are increased by voluntary physical activity and excitement and cease during sleep. Seizures are rare in adults with HD but affect approximately 50% of children. In some cases, they are the most prominent symptom and herald the onset of neurologic deterioration. Grand mal attacks are the most common; minor motor and myoclonic seizures and action myoclonus also have been observed (40). Spasticity, although rare in adults, can be prominent in juvenile HD (42). Cerebellar signs occasionally can be a major feature. Although optic nerve atrophy is not seen, impairment of ocular motility is an early sign and resembles oculomotor apraxia (9). In such instances, requesting the patient to look from side to side elicits closure of the eyelids or a rapid, jerking movement of the head with an attendant delay in the movement of the eyeballs. Unlike in oculomotor apraxia, random and vertical eye movements also are affected.
The disease progresses more rapidly in children than in adults, with an average duration of illness in the reported juvenile cases of 8 years, as contrasted with a range of 10 to 25 years for adults (43). Rate of progression of the disease has also been shown to be directly proportional to the number of CAG repeats.
Homozygotes for HD have been identified in a large Venezuelan kindred (44). Their neurologic picture and the age when symptoms became apparent did not differ from those of the heterozygotes, an indication that the HD gene is phenotypically completely penetrant in the heterozygous state.
Laboratory studies are normal, although the electroencephalogram result is often abnormal, and the cerebrospinal fluid (CSF) protein concentration is sometimes elevated. Computed tomography (CT) scanning and magnetic resonance imaging (MRI) are equally reliable in
demonstrating caudate atrophy, but can show normal results even when there are significant neurologic symptoms. The appearance of images on these scans is often not specific, but there can be abnormal T2 prolongation in the putamen (45). A more sensitive and more specific study involves positron emission tomography (PET) scanning after the infusion of radiolabeled fluorodeoxyglucose. A marked reduction in caudate glucose metabolism is observed in all symptomatic patients, regardless of whether atrophy is evident on CT or MRI scans, and in approximately one-third of presymptomatic individuals at risk for the disease (46). A reduced glucose metabolism is accompanied by a reduction in dopamine D1– and D2-receptor binding in caudate and putamen. A good correlation exists between intellectual deterioration and the reduction in D2-receptor binding (47).
demonstrating caudate atrophy, but can show normal results even when there are significant neurologic symptoms. The appearance of images on these scans is often not specific, but there can be abnormal T2 prolongation in the putamen (45). A more sensitive and more specific study involves positron emission tomography (PET) scanning after the infusion of radiolabeled fluorodeoxyglucose. A marked reduction in caudate glucose metabolism is observed in all symptomatic patients, regardless of whether atrophy is evident on CT or MRI scans, and in approximately one-third of presymptomatic individuals at risk for the disease (46). A reduced glucose metabolism is accompanied by a reduction in dopamine D1– and D2-receptor binding in caudate and putamen. A good correlation exists between intellectual deterioration and the reduction in D2-receptor binding (47).
Diagnosis
The capacity to monitor the size of the trinucleotide repeat in individuals at risk for HD has revolutionized preclinical and prenatal testing for the disorder. The following diagnostic guidelines have been established: (a) Allele sizes of 26 or fewer CAG repeats have never been associated with the HD phenotype or with mutations to longer repeats. (b) Allele sizes of 27 to 35 repeats have not been associated with the HD phenotype but have been associated with mutability when transmitted paternally. It is likely that some patients with sporadic HD represent the occasional expansion of high-normal paternal alleles into the affected range. (c) Allele sizes of 36 to 39 have been associated with HD phenotype but also have been seen in apparently unaffected individuals. (d) Allele sizes of 40 or more CAG repeats have always been accompanied by HD pathology (48). In the experience of Kremer and colleagues, who obtained molecular data from 1,007 individuals, 1.2% of patients with a clinical diagnosis of HD possessed repeats that fell into the normal range (49). It is not known whether these patients have a phenocopy of HD or whether they were misdiagnosed. Until effective treatment becomes available, the World Federation of Neurology does not advise that patients under 18 years of age be tested for the disease (50). I believe this is a complex issue and that the decision to administer or not to administer the test to children must be evaluated on a case-by-case basis.
The clinical diagnosis of HD rests on the neurologic examination, a positive family history, and neuroimaging studies. HD should be differentiated from the various conditions in which a movement disorder arises during childhood or adolescence. These include idiopathic torsion dystonia (dystonia musculorum deformans), Hallervorden-Spatz syndrome, Wilson disease, and juvenile Parkinson’s disease. When intellectual deterioration is the chief feature, the various lipidoses should also be considered. Since 1980 there has been a resurgence of Sydenham chorea in the United States. Even though an antecedent streptococcal infection usually cannot be elicited, the distribution of the involuntary movements assists in the diagnosis, as does immune evidence of a streptococcal infection.
Treatment
No treatment has been effective in reversing or arresting the progressive dementia. Choline, reserpine, and various phenothiazines, however, can alleviate the choreiform movements, and levodopa, bromocriptine, and amantadine can provide some benefit in the rigid form of the disease.
Huntington Disease–Like Disorders
There are several disorders that present with symptoms commensurate with HD but in which genetic analysis shows neither a pathologic CAG triplet expansion nor a linkage to 4p16.3. The best studied of these has been termed HD-like 2. It has been mapped to chromosome 16q24.4 and represents a CAG/CTG expansion in the gene that codes for junctophilin-3, a protein that is involved in the formation of junctional membrane structures (51). The clinical picture in this condition resembles that of HD.
In HD-like 3, a condition that has been mapped to chromosome 4p15.3, the clinical picture resembles that of juvenile HD, but transmission appears to be autosomal recessive (52). The condition manifests itself at about 3 to 4 years of age and is characterized by both pyramidal and extrapyramidal abnormalities. The MRI shows caudate atrophy.
HD-like 1 disease is linked to chromosome 20p12. This condition is a familial prion disease with a large insertion in the prior protein gene (53).
Hereditary benign chorea is a syndrome that is not too rarely encountered by a pediatric neurologist interested in movement disorders. This autosomal dominant condition, first described by Pincus and Chutorian (54), has its onset in infancy or early childhood and is nonprogressive, with peak severity in the second decade of life and movement diminishing during the adult years (55,56). Mild mental retardation is seen in a few patients, but there is no mental deterioration. PET scans in patients with this condition also demonstrate caudate hypometabolism. In some families the condition has been mapped to the long arm of chromosome 14, but there is significant genetic heterogeneity (57). Mutations in the thyroid transcription factor 1 gene, a homeodomain-containing transcription factor essential for the organogenesis of the lung, the thyroid, and the basal ganglia, have been documented in some patients carrying this disease (58).
Several conditions are now recognized in which an extrapyramidal disorder is accompanied by the presence of acanthocytes. These are red blood cells with an irregular body and spicules of variable length that are irregularly distributed over the surface of the cell (see Chapter 1,
Fig. 1.32). The differentiation between acanthocytes and red cells with rounded projections (echinocytes) and their visualization is discussed by Feinberg and coworkers (59). Acanthocytes are seen in more than 50% of all red cells derived from patients with abetalipoproteinemia but also have been observed in an occasional patient with Hallervorden-Spatz disease, HD-like 2 disease, hypobetalipoproteinemia, acanthocytosis, retinitis pigmentosa, pallidal degeneration syndrome (HARP syndrome: hypoprebetalipoproteinemia, acanthocytosis, and retinitis pigmentosa), and a condition termed chorea-acanthocytosis.
Fig. 1.32). The differentiation between acanthocytes and red cells with rounded projections (echinocytes) and their visualization is discussed by Feinberg and coworkers (59). Acanthocytes are seen in more than 50% of all red cells derived from patients with abetalipoproteinemia but also have been observed in an occasional patient with Hallervorden-Spatz disease, HD-like 2 disease, hypobetalipoproteinemia, acanthocytosis, retinitis pigmentosa, pallidal degeneration syndrome (HARP syndrome: hypoprebetalipoproteinemia, acanthocytosis, and retinitis pigmentosa), and a condition termed chorea-acanthocytosis.
Symptoms of HARP syndrome generally appear during the first decade of life and are marked by dystonia, dysarthria, and impaired gait (60).
Typically, chorea-acanthocytosis is characterized by chorea, orofacial dyskinesia with dysarthria, and depressed or absent reflexes. Seizures, dementia, and self-mutilation also have been observed. The age of onset is usually during the second decade or later (61). This condition is not rare in the south of Japan and can be transmitted in both a recessive and a dominant manner (62). The diagnosis of neuroacanthocytosis rests on the demonstration of acanthocytes in a dried blood smear or in saline-diluted wet preparations. Serum lipoprotein electrophoresis and vitamin E levels are normal. Neuroimaging demonstrates caudate atrophy, and MRI reveals increased signal in the caudate and lentiform nuclei. PET scans indicate a severe loss of striatal dopamine D2-receptor-binding sites and a marked reduction in the [18F]dopa uptake in the posterior putamen. These point to a selective degeneration of dopaminergic projections from the substantia nigra compacta to the posterior putamen (63). Further discussion of the various neurologic syndromes associated with peripheral acanthocytosis is given in Chapter 1.
Primary Dystonia (Dystonia Musculorum Deformans, Torsion Dystonia, DYT1)
Primary dystonia (dystonia musculorum deformans, torsion dystonia) is characterized by sustained muscle contraction frequently causing twisting and repetitive movements or abnormal postures. It was first described by Schwalbe in 1908 as a variant of hysteria (64) and more definitively by Oppenheim in 1911 (65). It is now evident that this condition represents several genetically distinct entities distinguishable from the various acquired dystonias that result from perinatal asphyxia, trauma, toxins, or vascular disease.
Pathogenesis and Molecular Genetics
Several genes have been implicated in the various hereditary dystonias. The disorders are summarized in Table 3.3. The clinical entity that most commonly manifests during childhood has been termed early-onset dystonia (DYT1). The gene for this condition has been mapped to chromosome 9q34. A single mutation, a 3-base pair (bp) deletion, in the coding sequence of the gene is found in almost all cases seen in Ashkenazi Jewish families (66). Identical gene mutations also have been documented in patients with completely different ethnicities. This suggests that DYT1 is an example of a recurrent mutation causing a dominantly inherited condition (85,86). The DYT1 gene codes for a protein termed torsin A, an adenosine triphosphate (ATP)-binding protein, which is a member of the AAA protein family. These proteins participate in a variety of biological functions including membrane trafficking, organelle biosynthesis, and protein chaperone activities. They achieve these tasks by unfolding proteins or disassembling protein complexes. The mutation causes torsin A to relocalize from the endoplasmic reticulum, where it is normally located, to the nuclear envelope, recruiting the wild-type torsin A to the nuclear envelope with it (87,88,89,89a).
The pathophysiology of primary dystonia (dystonia musculorum deformans) is unknown. Clinical and electrophysiologic observations indicate that dystonic movements and postures result from a functional disturbance of the basal ganglia. In particular, the striatal control of the globus pallidus and the substantia nigra pas reticulata is impaired. Microelectrode recording conducted in the course of pallidotomy shows a decreased discharge rate from both the internal and external segments of the globus pallidus, probably as a consequence of an increased inhibitory output from the striatum. In addition, the pattern of spontaneous neuronal activity is highly irregular (90). As a result, there is an alteration of thalamic control of cortical motor planning and execution of movements, reducing the brainstem and spinal cord inhibitory mechanisms (91,92). As a consequence, prolonged abnormal cocontractions of agonist and antagonist muscles occur, with abnormal recruitment (overflow) to the more distant musculature. Marsden postulated that a defect in the presynaptic inhibitory mechanisms in the spinal cord causes the failure in reciprocal inhibition (93).
Even though primary dystonia is believed to be a functional disorder of the basal ganglia, no consistent anatomic abnormalities are seen in this region. Neuropathologic examination of patients with dystonia musculorum deformans has been negative or has revealed nonspecific changes (94). However, McNaught and coworkers recently demonstrated the presence of perinuclear inclusions in cholinergic and other neurons of the midbrain reticular formation, an indication of altered protein handling such as is seen in several other neurodegenerative disorders (94a). Dopamine levels are slightly reduced in the rostral striatum, but not enough to explain the symptoms. This is in contrast to autopsy specimen from patients with
dopa-responsive dystonia, in whom dopamine levels range between 3% and 20% of normal (95). When discrete lesions result in a secondary focal or hemidystonia, these lesions are localized to the thalamus, caudate nucleus, putamen, or globus pallidus (93).
dopa-responsive dystonia, in whom dopamine levels range between 3% and 20% of normal (95). When discrete lesions result in a secondary focal or hemidystonia, these lesions are localized to the thalamus, caudate nucleus, putamen, or globus pallidus (93).
TABLE 3.3 Hereditary Dystonias | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Clinical Manifestations
Dystonia has been classified according to the body distribution of the abnormal movements. Generalized dystonia (dystonia musculorum deformans) is the most common form seen in children. In this condition, one or both legs and some other region of the body are affected (96). Focal dystonia refers to involvement of a single region. It includes the eyelids (blepharospasm), mouth (oromandibular dystonia), larynx (spastic dysphonia), neck (torticollis), and hand (writer’s cramp). In segmental dystonia, two or more contiguous regions are involved. Focal and segmental dystonias are seen rarely during the first two decades of life. Torticollis, when seen in infants and children, is a relatively common condition but is unrelated to primary dystonia. It is considered in Chapter 6.
DYT 1
Generalized DYT1 dystonia is transmitted as an autosomal dominant condition with a low penetrance, probably between 30% and 40% (97). The factors that control penetrance, be they genetic or environmental, are unknown (98). The degree of penetrance is not dependent on the gender of the transmitting parent.
Primary dystonia caused by a defect in the DYT1 gene is the most common type of childhood dystonia. It has a relatively high incidence among Ashkenazi Jews. This entity begins between the ages of 4 and 16 years and is characterized by an initially rapid deterioration. The first symptoms in both the childhood and late-onset forms of hereditary dystonia are involuntary movements or posturing of one portion of the body (99). Most commonly, the initial movement consists of a plantar, flexion-inversion movement of the foot that can first appear when walking (Fig. 3.2). Initially, symptoms are intermittent and, because they are more severe under stress, they frequently are misdiagnosed as representing a hysterical reaction. In due time, however, they become constant. As the disease progresses, involuntary flexion or extension appears at the wrist, and, finally, torsion spasms of the neck and trunk become evident. These movements can be triggered by motion of any other part of the body or ultimately they can appear spontaneously. Eventually, the affected limbs assume a fixed, continuously maintained, abnormal attitude on which athetotic fluctuations in time are superimposed. On examination, the muscles resist passive movements and after forcible deflection, tend to return to their original position.
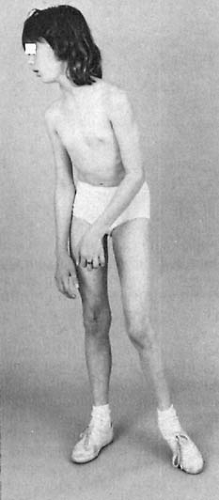 FIGURE 3.2. Dystonia musculorum deformans. Dystonic posturing of left arm and left lower extremity. Flexion at the knee and equinovarus positioning of the foot are typical for this condition. Additionally, this 11½-year-old girl has torticollis. Symptoms started at age 9 years with a change in voice, followed by torsion of the distal portion of her left lower extremity at age 10 years. |
In some cases, the involuntary movements affect the face and interfere with speech and swallowing. Dystonia of the visceral musculature, particularly paroxysmal dyspnea, has been described. As a rule, all movements cease during sleep. The intellect remains normal in all instances. The childhood-onset form of the disease tends to progress relentlessly, and, in general, the earlier the onset of symptoms, the more rapid is their advance. In the experience of Marsden and Harrison (100), 79% of children whose involuntary movements appeared before 11 years of age progressed to generalized dystonia. Individuals whose sole symptom is a mixed resting and intention tremor, writer’s cramp, or torticollis frequently are seen in affected families and probably represent a partial expression of the disease (99,101).
Neuroimaging in primary dystonia is usually unremarkable. On PET scan of a movement-free patient, a significant increase is seen in the cerebral glucose metabolism in the caudate and lentiform nuclei, cerebellum, and those areas of the frontal lobes that receive projections from the mediodorsal thalamic nucleus (92,102). In clinically nonpenetrant individuals, resting glucose utilization in putamen, anterior cingulate, and cerebellar hemispheres is also increased (103).
In contrast to the generalized forms of hereditary dystonia, segmental and focal forms are rarely encountered in children. The most common of these conditions is spasmodic torticollis, which can appear in isolation or as part of a segmental dystonia, with muscle spasms also affecting the upper extremity. Focal dystonias also include writer’s cramp, blepharospasm, spasmodic dysphonia, and oromandibular dystonia. Although these entities tend to remain restricted, they progress in some instances to generalized dystonia (104). In most instances these forms represent a partial expression of primary dystonia (DYT1) (101).
DYT5
A hereditary progressive form of dystonia with marked diurnal fluctuations and a striking response to low doses of dopaminergic drugs has been seen in Japan and accounts for approximately 5% to 10% of all dystonias seen in the West (71,105). Both autosomal dominant and autosomal recessive forms have been described (see Table 3.3). The
autosomal dominant form is the result of a defect in the gene that codes for guanosine triphosphate (GTP) cyclohydrolase I; the recessive form is caused by a defect in the gene for tyrosine hydroxylase (71,72). GTP cyclohydrolase I deficiency also is seen in hyperphenylalaninemia (see Chapter 1). The clinical picture for both forms is similar to early-onset generalized dystonia, with onset during the first decade of life. It generally startes with a postural dystonia in one extremity, and within a few years becomes generalized. A postural tremor of the hands is often present. In approximately one-fourth of children, the diurnal fluctuations are not apparent (71).
autosomal dominant form is the result of a defect in the gene that codes for guanosine triphosphate (GTP) cyclohydrolase I; the recessive form is caused by a defect in the gene for tyrosine hydroxylase (71,72). GTP cyclohydrolase I deficiency also is seen in hyperphenylalaninemia (see Chapter 1). The clinical picture for both forms is similar to early-onset generalized dystonia, with onset during the first decade of life. It generally startes with a postural dystonia in one extremity, and within a few years becomes generalized. A postural tremor of the hands is often present. In approximately one-fourth of children, the diurnal fluctuations are not apparent (71).
Patients with generalized dystonia and severe developmental delay and failure to achieve normal motor milestones also have been described. These patients are compound heterozygotes for mutations in the gene coding for GTP cyclohydrolase, but unlike patients with autosomal recessive GTP cyclohydrolase I deficiency, these patients do not demonstrate a marked elevation of plasma phenylalanine. These individuals respond dramatically to therapy with a combination of levodopa and tetrahydrobiopterin (BH4) (106). Diagnosis of this condition and related conditions such as the recently described infantile parkinsonism-dystonia depends on reliable quantification of the abnormal metabolites in the CSF (107,108).
DYT11
Myoclonic dystonia, a dominantly transmitted form in which torsion dystonia is accompanied by myoclonic jerks, can develop during childhood. Both myoclonus and dystonia improve dramatically with ingestion of alcohol or with a combination of trihexyphenidyl hydrochloride and valproic acid (109). The two genetically distinct forms appear to have similar phenotypes (Table 3.3).
Diagnosis
Primary dystonia (dystonia musculorum deformans) must be differentiated from dystonia arising as a consequence of a known central nervous system (CNS) insult (symptomatic or secondary dystonia) or because of other hereditary neurologic disorders. In children, dystonic symptoms are most often the result of perinatal asphyxia (see Chapter 6). Under such circumstances, the movement disorder generally appears before the third year of life, although, exceptionally, the involuntary movements do not begin until 5 to 15 years of age or even later, with the child having exhibited minimal or no neurologic abnormalities before that time (110,111). In a number of these patients, dystonic symptoms are not static but continue to progress throughout life.
Dystonic movements also can develop after acute viral encephalitis, carbon monoxide poisoning, and the ingestion of various drugs, notably phenytoin, carbamazepine, phenothiazines, butyrophenones, benzamides (e.g., Tigan), tricyclic antidepressants, chloroquine, antihistamines (e.g., Benadryl), ketamine, and lithium (112). Many children who develop drug-induced dystonia suffered from preexisting brain damage. Dystonia also can follow craniocerebral trauma. Under these circumstances, it is typically unilateral (hemidystonia). Typical dystonia also can follow peripheral trauma, with the traumatized limb being the one initially affected by the movement disorder (113,114). It is unclear what role trauma plays in the clinical expression that otherwise would have been asymptomatic for the DYT1 gene.
Dystonic movements also are seen in Leigh syndrome and Wilson disease. In Wilson disease, Kayser-Fleischer rings are invariable, the serum ceruloplasmin is generally depressed, and speech or the facial musculature is often preferentially affected. Other heredodegenerative diseases in which dystonia can be a prominent clinical feature include various lipidoses (GM1 and GM2 gangliosidosis, juvenile neuronal ceroid lipofuscinosis, and Niemann-Pick disease type C), metachromatic leukodystrophy (MLD), and glutaric acidemia type I.
Treatment
Two modes of treatment, pharmacologic agents and neurosurgery, have been used to alleviate the symptoms of dystonia. The effectiveness of drug treatment is unpredictable and inconsistent, but approximately one-half of the patients experience significant improvement. Trihexyphenidyl (Artane) is probably best used for DYT1 patients with this disease, whereas levodopa, given alone or with a decarboxylase inhibitor (Sinemet), appears to be most beneficial in patients with late-onset dystonia. Trihexyphenidyl is administered to children in extremely large doses (60 to 80 mg/day or even more). Doses begin at 2 to 4 mg/day, and the drug is gradually increased until maximum benefit or intolerable side effects are encountered (115). Tetrabenazine, a dopamine-depleting drug, can be given in doses of 50 to 200 mg/day. Its effect is potentiated by combining it with lithium (approximately 900 mg/day) (116). In some patients, a combination of trihexyphenidyl, dextroamphetamine, and tetrabenazine appears to be fairly effective, as is the cocktail of trihexyphenidyl, pimozide, and tetrabenazine used in the United Kingdom (117). The concomitant administration of baclofen (40 to 120 mg/day) can enhance the effectiveness of drug therapy (118). Other drugs that can be beneficial to an occasional patient include carbamazepine (Tegretol), bromocriptine, and diazepam (Valium).
Botulinum toxin (Botox) has been effective in the treatment of blepharospasm and other craniocervical dystonias, but it is used only rarely in generalized dystonia because of the number of large muscles affected. It can be used, however, for the most disabling or painful muscles in conjunction with the other medications (96).
Unilateral or bilateral pallidotomy and pallidal stimulation are options for patients who have been unresponsive to pharmacologic treatment. The latter procedure is preferred in our institution. Results from several centers indicate that this procedure induces a marked but gradual improvement over the course of several months (118a). Patients with secondary dystonia derive neither functional nor objective improvement (119,120). Pallidotomy appears to be equally effective (120). Because of a high incidence of relapses and the development of dysarthria in more than half of the patients who undergo bilateral procedures, cryothalamectomy is no longer used for amelioration of dystonia.
In general, it appears that not severely affected patients with the early onset form of hereditary dystonia (DYT1) respond more favorably to drugs, whereas patients with severe generalized DYT1 dystonia, and focal or segmental dystonia are likely to be more amenable to the use of botulinum toxin or surgery.
In light of the empirical and often not very successful treatment of patients with DYT1 dystonia, inhibiting the expression of the mutant torsin with the aid of mutant-specific small interfering RNA (siRNA) appears to be a new and potentially exciting approach (121).
Tardive Dyskinesia
Tardive dyskinesia is encountered in the pediatric population in association with the use of neuroleptic medications, notably phenothiazines, butyrophenones, and diphenylbutylpiperidine (Pimozide) (122). Its prevalence is much higher in children treated with conventional antipsychotics than in those given atypical antipsychotics. The condition is not one entity, but a number of different syndromes. Its incidence is not known. Studies on an inpatient population of psychotic children and adolescents receiving neuroleptics suggest that extrapyramidal movement disorders are seen during drug therapy or on drug withdrawal in as many as 50% (123). In most instances, the movement disorder is transient. It is more common in children who have had prenatal and perinatal complications or those who have received high doses of the medication for months or years or after an increase in the dose (124). However, low doses given over brief periods also have induced the movement disorder.
The severity of movements varies considerably. Characteristically, they consist of orofacial dyskinesias, notably stereotyped involuntary protrusion of the tongue, lip smacking, and chewing. A variety of other involuntary movements also can be present. After the medication is discontinued, these can improve in the course of several months or they can persist. Withdrawal dyskinesias generally disappear after a few weeks or months. Persistent dystonia has been associated with the use of neuroleptics. The clinical appearance of this movement disorder differs from tardive dyskinesia by the absence of orofacial dyskinesias.
Tardive dyskinesia is believed to result from a hypersensitivity of the dopamine receptors induced by the neuroleptic medications. A full review of the pathophysiology of this disorder is beyond the scope of this text.
Why this condition appears in some patients and not in others is unclear. The concomitant administration of anticholinergic drugs does not predispose to the appearance of tardive dyskinesia nor do drug holidays prevent it. In my experience, several patients with tardive dyskinesia have had a family history of a movement disorder (such as writer’s cramp or tremor), suggesting a genetic predisposition.
In making the diagnosis of tardive dyskinesia, one should bear in mind that a number of movement disorders, notably HD and Wilson disease, are preceded by behavior problems, personality changes, or psychoses that could require the use of neuroleptic agents. It is also necessary to differentiate tardive dyskinesia from the choreiform movements of hyperkinetic children, the peculiar movements of institutionalized youngsters exhibiting mental retardation, and the choreoathetosis of children with cerebral palsy that can be suppressed by neuroleptics.
A variety of drugs has been used for the treatment of persistent tardive dyskinesia. Jankovic and his group favor tetrabenazine given solely or in combination with lithium (116). In their series, which mainly comprised adults, a persistent excellent response was seen in 85% of patients. A variety of other drugs (reserpine, clonidine, and clozapine) also has been used. In many instances; however, if the movement disorder has established itself and has failed to subside with withdrawal of the responsible neuroleptic, the outlook for remission is not good. The effectiveness of branched-chain amino acids in the treatment of tardive dyskinesia is under investigation (125).
The neuroleptic malignant syndrome is another neurologic disorder believed to result from the administration of dopamine receptor–blocking neuroleptics. The principal clinical features of this condition are hyperthermia, muscular rigidity, tremors, rhabdomyolysis, and disorientation. Tachycardia and hypertension also can develop (126,127). The condition can be life-threatening, and prompt treatment is required. Anticholinergics, bromocriptine, and dantrolene have been used to treat this condition. Although the last drug is favored by many authors (128) it was not useful in the series of patients reported by Silva and coworkers (129).
Hallervorden-Spatz Disease (Pantothenate Kinase–Associated Neurodegeneration; PKAN)
In 1922, Hallervorden and Spatz described Hallervorden-Spatz disease, a rare familial disorder that began before age 10 years and was characterized by clubfoot deformity, gradually increasing stiffness of all limbs, impaired speech, and dementia (130).
Pathogenesis and Molecular Genetics
PKAN is an autosomal recessive disorder, with the gene mapped to chromosome 20p13. It encodes pantothenate kinase 2 (PANK2), the first enzyme in the biosynthesis of coenzyme A (131). Normally, PANK2 localizes to mitochondria and phosphorylates pantothenate, N-pantothenoyl-cysteine, and pantetheine (132). The mutated sequence leads to skipping of the mitochondrial targeting signal and causes PANK2 to be localized to the cytosol (133). It is not clear how this leads to the deposition of iron. One likelihood is that N-pantothenoyl-cysteine and pantetheine induce free radical damage to cells and that, additionally, iron is chelated by the cysteine-containing compounds that accumulate as the result of the enzyme defect.
Pathology
Three pathologic abnormalities occur in Hallervorden-Spatz disease. These are (a) intracellular and extracellular iron deposition in the globus pallidus, substantia nigra, and, less often, cerebral cortex; (b) a variable degree of neuronal loss and gliosis in the globus pallidus and the pars reticularis of the substantia nigra; and (c) widely disseminated rounded structures, identified as axonal spheroids and seen with particular frequency in the basal ganglia (134).
Microscopically, the pigment is located within neurons and glial cells and in the neuropil, particularly around large blood vessels of affected areas. Histochemical and chemical analyses indicate that it contains iron and is related to neuromelanin. The cytoplasm of the renal tubules contains a brown granular pigment similar in composition to that deposited in nerve tissues (135).
Clinical Manifestations
Considerable variability occurs in time of onset of symptoms and in the major manifestations of Hallervorden-Spatz disease (136). In the classic form of the disease, symptoms become apparent during early childhood (137). These include a progressive impairment of gait caused by a dystonic equinovarus pes deformity of the feet and rigidity of the legs, slowing and diminution of all voluntary movements, dysarthria, and in some children mental deterioration (138). Approximately 50% of patients, however, exhibit a choreiform and athetotic movement disorder. Spasticity has been evident in some cases and atrophy of distal musculature or seizures in others. Retinitis pigmentosa was seen in 20% of patients compiled by Dooling and colleagues (136) and in 68% of patients in the series of Hayflick and her group (137). Optic atrophy is seen in only about 3% of patients (137).
A form of PKAN that has a later onset and is more slowly progressive has also been delineated. Symptoms tend to be first noted in the second decade and are marked by speech difficulties and extrapyramidal dysfunction, the latter resembling parkinsonism (137).
Neuroimaging studies are of considerable assistance in the diagnosis of the disease. The CT scan shows bilateral high-density lesions within the globus pallidus (139). The presence of iron within the basal ganglia is best detected by MRI using a T2-weighted spin-echo pulse sequence at high field strength (1.5 Tesla). Marked hypointensity of the globus pallidus is seen, and the low-signal area surrounds a circumscribed region of high signal, the eye-of-the-tiger sign (140).
No effective treatment exists for the condition. In view of the enzymatic defect, high doses of pantothenic acid (up to 10 g/day) have been recommended in the hope of driving any residual enzyme activity by substrate overload (137). In my experience, neither pantothenic acid supplementation nor iron chelation has been effective. Movement disorders, notably dystonia, can sometimes be controlled by pallidal stimulation (140a). Pallidotomy has not been effective at our institution.
Familial Idiopathic Calcification of Basal Ganglia (Fahr Disease)
Fahr disease is not a single entity but several genetically and clinically distinct disorders that share the characteristic of calcific deposits within the basal ganglia (Fig. 3.3). A brain that demonstrated calcification within the basal ganglia was first described by Bamberger in 1855 (142). In 1930, Fahr reported a case of idiopathic calcification of the cerebral blood vessels in an adult with progressive neurologic symptoms (143). Since then, several genetic disorders subsumed under the term Fahr disease have been delineated (144).
One form of familial calcification of the basal ganglia is best exemplified by two sets of siblings reported by Melchior and coworkers (145). In these children, the disease was marked by mental deterioration, which became apparent within the first 2 years of life, and a choreoathetotic movement disorder. Microcephaly and seizures also can be present, and in one patient, calcification could be seen on ophthalmoscopic examination of the fundi. An autosomal recessive transmission is likely.
Another group has been designated as Aicardi-Goutiéres syndrome. It is an autosomal recessive progressive encephalopathy, with its onset in infancy, first described in 1984 (146). Initial symptoms include feeding difficulties and irritability. In some patients, previously acquired skills are lost and spasticity develops. Microcephaly is common (147). The diagnostic features include basal
ganglia calcifications, a mild but persistent CSF lymphocytosis, CSF protein elevation, and a high level of interferon-α in CSF and serum (147). MRI studies show increased signal in central white matter (148). In one variant of Aicardi-Goutiéres syndrome CSF interferon-α is normal but CSF neopterin and biopterin are elevated (149).
ganglia calcifications, a mild but persistent CSF lymphocytosis, CSF protein elevation, and a high level of interferon-α in CSF and serum (147). MRI studies show increased signal in central white matter (148). In one variant of Aicardi-Goutiéres syndrome CSF interferon-α is normal but CSF neopterin and biopterin are elevated (149).
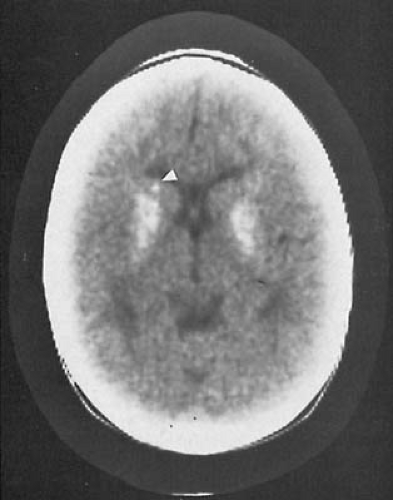 FIGURE 3.3. Calcified basal ganglia in a 10-year-old girl. Computed tomographic scan without contrast. The calcification is principally in the putamen and globus pallidus. Additionally, a focus of calcification in the caudate nucleus appears on the left (arrowhead). (Courtesy of Dr. Robert Sedgwick, and the late Dr. Lawrence S. Fishman, Children’s Hospital, Los Angeles, CA.) |
An autosomal dominant type of calcification of the basal ganglia is represented by the family reported by Boller and coworkers (150). Affected members developed chorea and dementia in the third or fourth decade. The family reported by Moskowitz and coworkers was clinically similar, and its members excreted greater-than-normal amounts of 3′,5′-AMP in response to administration of purified parathormone (151). In another family, calcification of the basal ganglia transmitted as an autosomal dominant trait was linked to chromosome 14q (152).
On pathologic examination, calcium deposits are found within capillaries and the media of the small and medium-sized arteries and less often the veins. Free calcium also is seen within brain tissue in the basal ganglia, cortex, and dentate nucleus of the cerebellum. There might be an accompanying demyelination (153).
Calcification of the basal ganglia also can be seen secondary to a number of well-delineated conditions. Most often, the picture is encountered in disorders of calcium metabolism, including hypoparathyroidism, pseudohypoparathyroidism, and hyperparathyroidism. Additionally, calcification of the basal ganglia, accompanied by deposition of the mineral in other parts of the brain, is seen in tuberous sclerosis, toxoplasmosis, cytomegalic inclusion disease, AIDS, chronic Epstein-Barr infection, Cockayne syndrome, Wilson disease, and Down syndrome (154). Calcification of the basal ganglia has variously been reported as a consequence of perinatal asphyxia, but in my experience, this situation is extremely unusual. A differential diagnosis of basal ganglia calcification as detected by CT scans was presented by Harrington and coworkers (155).
Hereditary (Familial) Tremor
Hereditary (familial) tremor represents the most common yet the most benign movement disorder seen in childhood. Symptoms usually become evident around puberty, although patients aged 5 years or younger have been observed.
The condition is polygenic, and in most families it is transmitted as an autosomal dominant trait. Two genetic loci have been established. One, obtained from an Icelandic family, is on chromosome 3q13 (FET1) (156). The other locus was obtained from a large American family and is on chromosome 2p22–p25 (FET2) (157). The presence of clinical anticipation suggests that an expanded CAG trinucleotide sequence will turn out to be responsible for the disease in some families. Without doubt, other gene loci will be found in the course of the next few years.
The pathophysiology of familial tremor is in dispute. Autopsy has shown no significant abnormality (157a). Marsden distinguished two forms. The first he termed an exaggerated physiologic essential tremor with a frequency of 8 to 12 Hz, which depends on a peripheral reflex. The second form represents a pathologic essential tremor having a frequency of 4 to 7 Hz, which is largely dependent on an interaction between an overactive central generator, which does not arise in the cerebellum, but is transmitted through the cerebellothalamocortical pathway. It arises by abnormal cerebellar afferent input from areas such as the inferior olive (158,159). On PET, overactivity of this loop is confirmed by the observed hypermetabolism of the medulla, presumably reflecting inferior olivary activity, and abnormal bilateral cerebellar blood flow during tremor and at rest (160).
Essential tremor in childhood is seen predominantly in boys (161). On examination, there is a rhythmic tremor, with the frequency of oscillation usually being between 4 and 8 Hz (158). The tremor increases with movement, becoming accentuated as the limb approaches its target, and, except in the most severe cases, is absent when the limb is at rest. It disappears with sleep. The tremor initially affects one or both of the upper extremities but usually does not interfere with fine coordination. With tremor progression, the patient can develop head nodding. The lower extremities are affected less often, and intelligence, gait, and strength remain normal (162,163). Speech and eyelids are rarely affected during childhood, and head tremor
is unusual. Brief episodes of shuddering or stiffening of the body, usually associated with excitement, might be observed in infancy or early childhood. They are believed to represent an antecedent to familial tremor (164).
is unusual. Brief episodes of shuddering or stiffening of the body, usually associated with excitement, might be observed in infancy or early childhood. They are believed to represent an antecedent to familial tremor (164).
Although the disease is progressive during its early stages, it arrests during much of adult life and interferes little with the patient’s daily activity. In later life, however, the condition can suddenly become aggravated, and senile tremor can be a late manifestation of essential tremor.
The diagnosis of essential tremor rests on the exclusion of other disorders of the basal ganglia. Tremors can accompany DYT1 (162), spasmodic torticollis, and writer’s cramp. They probably represent a partial expression of primary dystonia. Tremors also are seen in Wilson disease and HD. A family history of the disorder and examination of the parents and other family members are helpful.
Treatment
In most cases of familial tremor, the tremor is not sufficiently severe to require treatment during childhood and adolescence. Propranolol, a nonselective β-adrenoreceptor antagonist, relieves familial tremor. It also has been used to treat shuddering attacks (165). For adults, doses range from 120 to 240 mg/day, given in divided doses. No correlation exists between plasma levels of propranolol and tremorolytic action (166). Small doses of primidone (50 mg/day) or of topiramate (50 mg/day) are equally and in some patients even more effective (167,168).
Restless Legs Syndrome
Restless legs syndrome is a common disorder. It affects between 2% and 5% of the population. It is characterized by a creeping-crawling or aching sense of discomfort in both legs that creates an irresistable need to move them. Symptoms are most common at night before going to sleep. In addition, brief involuntary flexion of the lower extremities can be noted during sleep. Occasionally, the movements also affect the upper extremities. In approximately one-third of patients, there is a family history, and an autosomal dominant transmission appears likely. The cause for the movements is not clear. Trenkwalder and colleagues suggested the presence of brainstem disinhibition that activates a spinal generator for the involuntary limb movements (169). CSF orexin-A levels are increased in patients, most strikingly in those with an early onset of the condition (169a). In the experience of Walters and colleagues, approximately 20% of adult patients with restless legs syndrome had an age of onset before 10 years. Diagnosis was usually not made at that time; instead, the condition was considered as growing pains or attention-deficit/hyperactivity disorder (ADHD) (170). The association of ADHD and restless legs syndrome has also been made prospectively. In the series of Picchietti and Walters, 15 of 16 children with severe restless legs syndrome also had ADHD (171). The diagnosis of restless legs syndrome is best made by polysomnography or videotaping, which discloses the periodic limb movements during sleep. Treatment with clonazepam or adopaminergic agent such as levodopa or bromocriptine has been quite effective (172).
Juvenile Parkinson’s Disease
As is commonly defined, juvenile Parkinson’s disease refers to a condition in which symptoms of Parkinson’s disease make their appearance before age 20 years. This is in distinction to early-onset Parkinson’s disease, a condition that becomes apparent between the ages of 20 and 40 years. Our concepts of juvenile Parkinson’s disease have undergone marked changes. Although it had been known for many years that when Parkinson’s disease develops during the first two decades of life, the familial occurrence can be as high as 40%, the genetic transmission had not been clarified until recently.
The initial report of juvenile Parkinson’s disease by Hunt in 1917 probably described four unrelated adolescent patients with dopa-responsive dystonia (173). Since then, genetic analysis has confirmed the clinical impression that juvenile Parkinson’s disease is a heterogeneous condition, and at least three autosomal dominant and at least three autosomal recessive forms have been delineated.
One of the genes for an autosomal dominant form has been mapped to chromosome 4q21–q23 (PARK1) (174). This gene encodes α-synuclein, a neuron-specific presynaptic membrane protein, which is a component of Lewy bodies, the fibrous cytoplasmic inclusions seen in dopaminergic neurons of the substantia nigra in adult-onset Parkinson’s disease (175). Two mutations in the gene for synuclein have been described (176).
Other loci for autosomal dominant early-onset Parkinson’s disease (PARK3) have been mapped to chromosome 2p13 (177), and chromosome 12q12 (177a). The genes for the autosomal recessive forms of juvenile Parkinson’s disease (PARK2, PARK6, PARK7) have been mapped to chromosomes 6q15.2–q27, 1p35–p36, and 1p36, respectively (178). The gene for PARK2 encodes a protein, named parkin, which is abundant in all parts of the brain, including the substantia nigra (179). A variety of mutations in the gene, including deletions and point mutations, can lead to clinical picture of juvenile Parkinson’s disease (180,181).
The clinical picture of PARK1 and PARK3 is similar, and in most instances symptoms do not become apparent until the third or fourth decade. Autosomal recessive PARK2 is encountered frequently in Japan (182). Symptoms appear as early as the second half of the first decade of life. Initial signs are a gait disturbance with retropulsion, a fine tremor, hyperreflexia, and dystonic posturing of the foot. Parkinsonian symptoms improve with sleep. Good response occurs with levodopa, although dopa-induced dyskinesia can appear within a few months of starting
the medication. No intellectual deterioration occurs. MRI study results are normal (182). Neuropathologic studies show neuronal loss and gliosis in the pars compacta of the substantia nigra. In the median portion of the substantia nigra and in the locus ceruleus, neuronal loss and poor melanization of the nigral neurons occur. No Lewy bodies are detectable in either the autosomal recessive or the autosomal dominant forms (183,184).
the medication. No intellectual deterioration occurs. MRI study results are normal (182). Neuropathologic studies show neuronal loss and gliosis in the pars compacta of the substantia nigra. In the median portion of the substantia nigra and in the locus ceruleus, neuronal loss and poor melanization of the nigral neurons occur. No Lewy bodies are detectable in either the autosomal recessive or the autosomal dominant forms (183,184).
The pathophysiology of Parkinson disease is a topic that should be relegated to a text on adult disorders.
Juvenile Parkinson’s disease should be distinguished from Wilson disease, dopa-responsive dystonia, and spinocerebellar ataxia 2 (SCA2) or other spinocerebellar ataxias that can manifest with prominent parkinsonism. We have encountered identical twins with a clinical picture of the dystonic, dopa-responsive form of juvenile Parkinson’s disease who were found to have neuronal intranuclear inclusion disease (185). Another condition to be considered is rapid-onset dystonia-parkinsonism (DYT12), which can have its onset as early as the second decade of life (80,186).
Familial Infantile Bilateral Striatal Necrosis
This rare entity, first described by Paterson and Carmichael (187), is probably an autosomal recessive condition and has been mapped to chromosome 19q (188). It is marked by developmental arrest, dysphagia, choreoathetosis, pendular nystagmus, and optic atrophy. MR imaging studies demonstrate marked atrophy of the basal ganglia. On autopsy there is severe atrophy of the lenticular nuclei with neuronal loss and gliosis (189). Most often the condition develops gradually, but there have been instances in which it appeared acutely following an infectious illness. It is not clear whether this acute form and another form that develops later in life are expressions of the same genetic defect.
Gilles de la Tourette Syndrome
Gilles de la Tourette syndrome, which straddles the fields of neurology and psychiatry, was first described in 1885 by Georges Gilles de la Tourette (190). It is characterized by multiple, uncontrollable muscular and vocal tics and, in the classic cases, obscene utterances. Although in the past the condition was considered to be rare and caused by emotional and psychiatric factors, it is now clear that it represents a continuum from mild and transient tics at one end to the complete Tourette syndrome (TS) at the other.
Pathogenesis and Pathology
The pathogenesis of TS is unknown. Although the current working hypothesis proposes an autosomal dominant transmission of the disease, this has not been fully documented. Family studies have shown that the increase of TS in first-degree relatives of TS patients is at least beween 10% and 15%, a value that is about 10 times that seen in the general population (191). There also is a significant difference in concordance for monozygotic and dizygotic twins (191). A systematic genome scan has shown that four regions (4q, 7q, 8p, and 11q) may contain TS-related genes (192). Specifically, no confirmed linkage has been documented to the various dopamine-receptor genes or to the dopamine-transporter gene. Failure to establish such a linkage could reflect diagnostic uncertainties, including an unclear definition of TS, the variable clinical expression of the disorder, and genetic heterogeneity. Walkup and coworkers suggested that the susceptibility for TS is inherited by a major locus in combination with a multifactorial background (193). Because cases of secondary TS, such as after carbon monoxide poisoning, have shown lesions in the basal ganglia (194), a disorder in the circuitry of the basal ganglia has been postulated, with a working hypothesis being that TS is a developmental disorder of synaptic transmission that results in the disinhibition of the cortico-striatal-thalamic-cortical circuitry (192,195).
Neuropathologic studies have failed to find any consistent structural alterations within basal ganglia. Volumetric MRI suggests that the left-sided predominance of the putamen visualized in healthy right-handed boys is absent in TS and that there is a tendency to right-sided predominance (196). Neurochemical examinations indicate low levels of serotonin in brainstem, low levels of glutamate in globus pallidus, and low levels of cyclic AMP in the cortex (192). Using single-photon emission computed tomography or PET, some but not all studies have found an increase in the density of the presynaptic dopamine transporter and the postsynpatic D2 dopamine receptor, a suggestion that there is abnormal regulation of dopamine release and uptake. These findings have been partly confirmed by postmortem studies that demonstrate increased prefrontal (but not striatal) D2 protein in TS brain (197).
The observation that children with TS have significantly increased antineuronal antibodies against human putamen suggests an autoimmune mechanism. A number of studies have found a relationship between a prior streptococcal infection and various movement disorders aside from Sydenham chorea, but such a link has not been proven for TS (197a). This topic is covered more fully in Chapter 8 in the section on pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS).
Clinical Manifestations
A variety of estimates have been presented for the prevalence of TS in the United States. These range from 5 in 10,000, as determined in North Dakota children (198), to as high as 4.2% when all types of tic disorders are included (192). Boys are affected more frequently than girls by a
ratio of between 3:1 and 5:1. The syndrome is rare in blacks; in Cleveland, 97% of TS patients were white (199). In approximately 33%, the condition is familial, and relatives can have multiple tics, an obsessive–compulsive disorder, or the complete TS (200,201).
ratio of between 3:1 and 5:1. The syndrome is rare in blacks; in Cleveland, 97% of TS patients were white (199). In approximately 33%, the condition is familial, and relatives can have multiple tics, an obsessive–compulsive disorder, or the complete TS (200,201).
Symptoms generally appear between ages 5 and 10 years. Although periods of remission occur and TS tends to become less severe after adolescence, the condition usually persists for life, and only 8% of children have complete and permanent remission. In the survey of Pappert and colleagues more than one-fourth of adults with TS were disabled due to alcohol abuse, criminality, and unemployment. Tic severity was not, however, a primary factor for disability. It was of note that early life dysfunction correlated with dysfunction during adult life (202).
The initial clinical picture is usually one of multifocal tics affecting the face and head. These progress to vocal tics, such as grunting, coughing, sneezing, and barking and, in the most severe cases, to coprolalia or compulsive echolalia. Tics can be voluntarily suppressed for variable periods, but an accompanying increase in internal tension ultimately results in a symptomatic discharge. Compulsive self-mutilation has been noted in a significant number of cases. In patients in the Cleveland series, an attention-deficit disorder was documented in 35%, learning disabilities in another 22%, and serious psychiatric disorders in 9% (197a). Coprolalia was encountered in only 8%, an indication that even mild cases of the syndrome are coming to physicians’ attention. An increased incidence in TS during the winter months has been observed, and the role of group A streptococcal infection as a trigger for tics has been suggested by several workers (203,204). As observed in the genetic studies, obsessive–compulsive traits also are characteristic; in contrast to TS they tend to occur more frequently in girls (200).
Clinical findings suggest that brain function is abnormal in a high proportion of these children. On neurologic examination, a large number of patients exhibit “soft” signs. Approximately 15% of cases are precipitated by stimulant medications, such as methylphenidate hydrochloride (Ritalin), pemoline (Cylert), and dextroamphetamine sulfate (Dexedrine) (205).
Electroencephalogram results have been reported to be abnormal in as many as 55% of affected patients, sometimes even revealing frank paroxysmal discharges. Sleep is disturbed. The amount of stages 3 and 4 sleep is increased, the number of awakenings is increased, and the amount of rapid eye movement sleep is decreased. Sleep electroencephalography (EEG) shows high-voltage slow waves with awakening and immature arousal pattern (206).
Treatment
The goal of treatment is to achieve a satisfactory balance between maximal control of tics and a minimum of side effects. Several medications are being used, but few have been tested in placebo-controlled studies (191). Postsynaptic D2-receptor antagonists are generally the most reliable drugs for the treatment of TS. Although a recent publication lists 20 drugs that have been advocated for treatment, most clinicians favor haloperidol or pimozide (206a). Haloperidol (0.5 to 2.0 mg/day) improves symptoms in up to 80% of affected children, although side effects prevent the drug from being used for long-term therapy in more than one-third of patients (207), and Robertson and Stern no longer consider it as the first-line drug (208). Because of the marked sedation and alterations in personality induced by the drug, I agree with them. Pimozide appears to be at least as effective as haloperidol, producing significant improvement in 70% of patients with fewer side effects (209). As recommended by Shapiro and coworkers, the starting dose for children is 1 mg given at bedtime, with the dose increased 1 mg every 5 to 7 days until at least a 70% reduction of symptoms occurs or until adverse side effects appear (209). Regeur and coworkers used a dose range of 0.5 to 9.0 mg/day (210). Pergolide, a mixed D1, D2, D3 agonist, was used by Gilbert and coworkers at Cincinnati Children’s Hospital with good results in a randomized, double-blind crossover trial, but other centers have not found it to be as efficacious (211). Kurlan considers α-adrenergic agonists, such as clonidine or guanfacine, to be the first-line medications for the treatment of tics, with second-line agents being the atypical antipsychotics such as resperidone and olanzapine (212).
Other drugs that have been advocated for the treatment of TS include tetrabenazine, clonidine, and fluphenazine. The variety of medications that have been proposed indicates that none are of exceptional value and that all have their drawbacks.
A large proportion of children with TS (21% to 90%) develop symptoms of ADHD at some time in their life. Because many of the medications used in the treatment of TS are sedative, they tend to worsen ADHD, and generally ADHD needs to be treated as well. In my practice I find that given the choice, it is more important for the emotional adjustment of the child and the family to treat the ADHD than the TS. Kurlan and colleagues found that methylphenidate and clonidine particularly when given in combination are effective for children with ADHD who have comorbid tics or TS. In their series of children with combined TS and ADHD, methylphenidate used in the same dose as in children without tics was quite effective, and tics worsened in only 20% of patients, a figure that was no greater than in placebo-treated children (213). I should point out that Leckman and coworkers found that clonidine might not become effective until it has been given for some 3 months. They also noted that even after brief discontinuation, its reinstitution does not result in an improvement until after 2 weeks to 4 months (214).
Side effects are similar for all drugs but are least common with pimozide. Acute dystonia is seen in some 5% to 9% of patients receiving pimozide; akathisia is seen in
some 25%, but usually disappears within the first 3 months of therapy. Tardive dyskinesia is seen with haloperidol and pimozide; it is more common with haloperidol (215). Other side effects include weight gain, gynecomastia, and, rarely, nonspecific alterations of the electrocardiogram.
some 25%, but usually disappears within the first 3 months of therapy. Tardive dyskinesia is seen with haloperidol and pimozide; it is more common with haloperidol (215). Other side effects include weight gain, gynecomastia, and, rarely, nonspecific alterations of the electrocardiogram.
Paroxysmal Dyskinesias
The first entity of this rare set of disorders, which straddle the boundary between seizures and basal ganglia diseases, was described by Mount and Reback in 1940 (216). In its classic form, it is characterized by paroxysmal attacks of choreiform, athetoid, dystonic, or tonic movements lasting from a few seconds to several minutes. Consciousness remains unimpaired. Goodenough and coworkers (217) classified the reported cases into familial and acquired (sporadic), with the majority falling into the latter group (218).
Lance distinguished two types of familial dyskinesias (219). In one group, termed kinesigenic paroxysmal dyskinesias (PKDs, DYT10), attacks are regularly precipitated by movement, especially movement occurring after prolonged immobility. Paroxysms begin almost always in childhood, usually between ages 6 and 16 years. The movements most often resemble dystonia, although some patients can exhibit a choreiform or even a ballistic component (77). About one-fourth of patients in the series of Houser and coworkers had other affected family members (220). Attacks usually last less than 5 minutes. In the familial cases, the condition appears to be transmitted as an autosomal dominant trait. Attacks respond promptly to anticonvulsants, notably carbamazepine or phenytoin (220). Curiously, blood levels required to control attacks (4 to 5 μg/mL) are well below the range considered to be therapeutic for seizure disorders (221).
The condition has been mapped to 16p11.2–q12.1 (222). In several families there is an association between PKD and benign infantile convulsions (224).
The pathophysiology of this disorder is unclear. Mir and coworkers have recently found characteristic abnormalities in cortical and spinal inhibitory circuits, in particular a reduced short intracortical inhibition (223). In my experience paroxysmal nonkinesigenic dykinesia (PNKD, DYT8) is a more common entity. Attacks arise spontaneously or are triggered by fatigue. They first appear before 5 years of age and most often take the form of dystonia, although a mixed movement disorder also can be seen (220). The family history is consistent with an autosomal dominant transmission, and the gene has been mapped to chromosome 2q34 (225). In the experience of Demirkiran and Jankovic, attacks usually last longer than 5 minutes in 65% of instances, but the duration of attacks cannot be used to classify the various paroxysmal dyskinesias (220). The response to anticonvulsants is not as good as in the kinesigenic cases. Between attacks, the child is usually in excellent health, although some children have shown mild choreiform movements or a seizure disorder and some have been developmentally delayed (226,227).
Another autosomal dominant PNKD has been mapped to chromosome 1p (228). In this condition, a spastic paraplegia occurs during and between episodes of dyskinesia. Less often one encounters exertion-induced dyskinesias or sleep-induced dyskinesias (220).
Acquired paroxysmal dyskinesias are often seen in children who had previously experienced perinatal asphyxia or other static encephalopathies as well as in patients with reflex epilepsy and metabolic disorders, notably idiopathic hyperparathyroidism. The condition also is seen in multiple sclerosis. In some cases of acquired paroxysmal dyskinesia, attacks have responded to diphenhydramine (227). Other patients respond to clonazepam or acetazolamide. Bressman and coworkers distinguished a psychogenic form of paroxysmal dystonia. Patients belonging to this group respond to placebo, hypnotherapy, or faith healing (218).
A transient form of dystonia arising during infancy is marked by periods of opisthotonos and dystonic posturing of the upper limbs occurring during the waking state and generally lasting no more than a few minutes. The attacks resolve within a few months. No family history exists, and this entity is distinct from Sandifer syndrome, which accompanies hiatus hernia (see Chapter 6), and from paroxysmal torticollis, a precursor to migraine (229).
The essence of the paroxysmal dyskinesias is unknown. Stevens (230) and Homan and associates (222) postulated that they represent a convulsive disorder akin to the reflex epilepsies. During an attack, however, the EEG does not disclose any paroxysmal features and differs little from the interictal tracing. Most likely, in analogy with paroxysmal ataxia, a condition covered in a subsequent section, the paroxysmal dyskinesias will turn out to represent different types of channelopathies.
Hyperekplexia
Familial startle disease or hyperekplexia is characterized by transient, generalized rigidity in response to unexpected loud noises or sudden tactile stimulation. A large number of patients exhibit severe muscle stiffness at birth (stiff baby syndrome) (231). Infants present with marked irritability and recurrent startles in response to handling or sounds, or tapping on the face or the bridge of the nose. This can be accompanied by rhythmic, jerky movements and occasionally by episodes of breath holding (232). The condition is misdiagnosed frequently as spastic quadriparesis. Muscle tone returns to normal in early childhood, but patients continue to have sudden falls in response to startle and they can suppress the startle reaction if they are able to anticipate the stimulus (233). The “jumping Frenchmen of Maine” probably represent an extensive family with such a condition (234). The EEG and electromyography (EMG) results are generally normal.
The condition has been mapped to chromosome 5q33–q35 (235), and in most instances it is caused by mutations in the alpha1 subunit (GLRA1) of the inhibitory glycine receptor. This receptor facilitates fast-response, inhibitory glycinergic neurotransmission in the brainstem and spinal cord leading to a reduction of the excitatory startle response. Both autosomal dominant and recessive forms have been described, arising from different mutations of GLRA1 (236,237,238). Mutations in the beta subunit of the inhibitory glycine receptor have also been found (239). Symptoms improve markedly with administration of clonazepam or valproate.
HEREDODEGENERATIVE DISEASES OF THE CEREBELLUM, BRAINSTEM, AND SPINAL CORD
Heredodegenerative diseases of the cerebellum, brainstem, and spinal cord involve a slow, progressive deterioration of one or more of the functions subserved by the cerebellum, brainstem, or spinal cord. This deterioration is caused by neuronal atrophy within the affected tract or tracts, commencing at the axonal periphery and advancing in a centripetal manner. Familial distribution is the usual pattern. In the past, classification of these disorders based on clinical and pathologic data led to numerous debates, which are now being resolved with the increasing availability of genetic data. Two groups can be distinguished: the autosomal recessive ataxias and the autosomal dominant ataxias. In this chapter, emphasis is placed on those conditions that are more prevalent during childhood and adolescence.
Friedreich Ataxia
Friedreich ataxia (FRDA) was first distinguished from syphilitic locomotor ataxia by Friedreich in 1863, who reported all the essential clinical and pathologic features of the condition (242). Characterized by ataxia, nystagmus, kyphoscoliosis, and pes cavus, it occurs throughout the world as an autosomal recessive trait.
Molecular Genetics, Pathology, and Pathogenesis
The gene for FRDA has been mapped to chromosome 9q13. It has been cloned and codes for a protein termed frataxin (243). Some 94% of FRDA patients are homozygous for an expanded GAA repeat in an intron of the gene. The remaining individuals, some 5%, are compound heterozygotes with an expanded repeat on one allele and a point mutation or deletion on the other (244). Compound heterozygotes have a somewhat earlier onset of symptoms and are more likely to show optic atrophy (245). The number of GAA repeats ranges from 6 to 29 in healthy individuals and from 120 to 1,700 in FRDA patients, with the size of the expanded repeat correlating fairly well wth age of onset and disease severity (244,246,247). The expression of frataxin is markedly reduced in FRDA, and the pattern of frataxin expression in normal tissues approximates the sites of pathologic changes in the disease (243). Frataxin is normally localized to mitochondria, where it plays a critical role in iron homeostasis (248). In yeast and mouse models of FRDA frataxin deficit results in reduced activity of numerous mitochondrial enzymes, notable complexes I, II, and III of the mitochondrial respiratory chain (249), and aconitase (250). Impaired mitochondrial function is probably the result of iron overload in mitochondria and tissue damage from free radical toxicity and abnormal antioxidant responses (251,252). In patients with FRDA there is an accumulation of iron in myocardial cells and in the dentate nucleus (244). The mechanism by which frataxin regulates iron metabolism is uncertain, but it is likely that the protein acts as an iron chaperone or an iron store (248,250). How the GAA triplet-repeat expansion results in a reduced expression of frataxin in mitochondria is not clear. The most likely possibility is that the unusual DNA structure interferes with transcription (253). Interestingly, the at one time discredited finding of abnormalities in the pyruvate dehydrogenase complex and in the activity of the mitochondrial malic enzyme can now be explained (254).
The essential pathologic process in FRDA as well as in the other cerebellar degenerations is a dying back of neurons in certain systems from the periphery to the center with eventual disappearance of the cell body (255). The principal lesions are found in the long ascending and descending tracts of the spinal cord (Fig. 3.4). The peripheral nerves also can be affected, as can the brainstem and, less often, the cerebellum. All these areas show axonal degeneration, demyelination, and a compensatory gliosis. Degenerative cellular changes are most striking in Clarke column and the dentate nucleus. They occur to a lesser extent in a variety of other nuclei of the lower brainstem and in the Purkinje cells. Cell loss and gliosis also are seen in the vestibular and auditory systems (256). Other areas of gray matter are usually unaffected.
Abnormalities of the viscera are limited to the heart and the pancreas. There is cardiomegaly, with myocytic hypertrophy and chronic interstitial fibrosis, and an inflammatory infiltrate (257). In the pancreas there is a loss of islet cells.
Clinical Manifestations
Friedreich ataxia accounts for about three-fourth of cases in whites with hereditary ataxia having their onset before age 25 years. The prevalence in Great Britain is
approximately 1 in 48,000 (257). In western Norway, it is 1 in 100,000. Although considerable variability occurs in age of onset, in most families the disease begins at the same period of time in each affected individual. The first symptoms can be seen as early as age 2 years, and the mean age of onset is 10 years of age (258). The clinical manifestations are outlined in Table 3.4 (259). Children can be slow in learning to walk or can begin to stumble frequently. Less commonly, abnormal speech or incoordination of hand movements is the initial complaint.
approximately 1 in 48,000 (257). In western Norway, it is 1 in 100,000. Although considerable variability occurs in age of onset, in most families the disease begins at the same period of time in each affected individual. The first symptoms can be seen as early as age 2 years, and the mean age of onset is 10 years of age (258). The clinical manifestations are outlined in Table 3.4 (259). Children can be slow in learning to walk or can begin to stumble frequently. Less commonly, abnormal speech or incoordination of hand movements is the initial complaint.
 FIGURE 3.4. Friedreich ataxia. Section of spinal cord stained for myelin. The area of myelination involves the posterior columns and the lateral and ventral spinocerebellar tracts. The corticospinal tracts are relatively spared, although demyelination is more extensive than would be expected from the clinical picture at the time of demise. (Courtesy of Dr. K. E. Earle, Armed Forces Institute of Pathology, Washington, DC.) |
TABLE 3.4 Frequency of Neurologic Symptoms in 72 Patients with Friedreich Ataxia | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Neurologic symptoms advance relentlessly. Although the disease evolves rapidly in a few children, progression is slow in the vast majority, and, occasionally, long static periods occur. Intercurrent infections frequently aggravate existing symptoms and bring new ones to light. The patient with fully developed FRDA has an immature, dysmorphic appearance with a number of skeletal deformities, some of which can exist from birth. In approximately three-fourths of the patients, the feet have a high arch (pes cavus), hammer toes, and wasting of the small muscles of the sole (Fig. 3.5). This abnormality, present at birth, might for many years represent the only sign of FRDA. Kyphoscoliosis is present in 75% to 90% of patients (257,259).
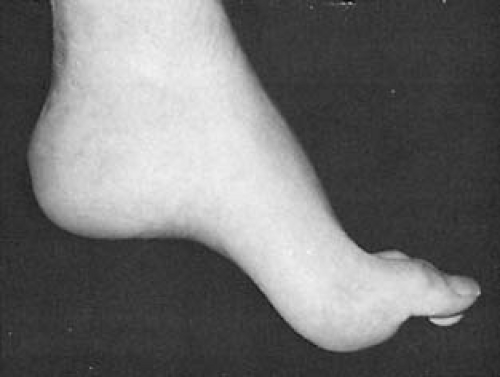 FIGURE 3.5. Typical pes cavus foot deformity seen in Friedreich ataxia. The patient is a 12-year-old girl. |
The most prominent sign, however, is ataxia. As might be expected from the pathologic lesions, ataxia is caused by a combination of cerebellar asynergia and loss of posterior column sensation. Ataxia is usually more marked in the legs than in the arms and is most evident when the child’s gait or station is examined. Speech is invariably involved and acquires a staccato or explosive character, which is the result of an incoordination of respiration and phonation. Nystagmus is seen in 20% to 40% of cases (257,258,259). It is usually bilateral and is present only on lateral movements of the eyes. Additionally, one frequently observes broken-up, jerky pursuit movements. Nystagmus on upward gaze is rare, as is optic atrophy (255). In the series of Pandolfo (260) the latter was seen in 13% of cases. Optic atrophy can be congenital or can have its onset during early infancy, progressing rapidly after its inception.
Carroll and coworkers found color vision to be impaired in 40% of patients, and abnormalities in the visual-evoked potentials were seen in two-thirds of the patients (261). The two principal abnormalities were a generalized reduction in amplitude of the potentials and a prolongation in their latency. The reduction in amplitude is probably the consequence of a loss of functional fibers in the
visual pathway. Electroretinogram results are usually normal. Vestibular involvement, including loss of caloric reactions and attacks of vertigo, was already described by Friedreich (242). Attacks of vertigo often appear early and ultimately are seen in approximately 50% of the patients. Sensorineural hearing loss was seen in about 20% of Pandolfo’s series; it is caused by degeneration of the cochlear neurons (256,260,262).
visual pathway. Electroretinogram results are usually normal. Vestibular involvement, including loss of caloric reactions and attacks of vertigo, was already described by Friedreich (242). Attacks of vertigo often appear early and ultimately are seen in approximately 50% of the patients. Sensorineural hearing loss was seen in about 20% of Pandolfo’s series; it is caused by degeneration of the cochlear neurons (256,260,262).
Weakness of the distal musculature of the lower limbs and wasting of the small muscles of the hands and feet are common and are out of proportion to the degree of disuse. A loss of vibratory and proprioceptive sensation occurs, and in the advanced cases, the other sensory modalities also are likely to be affected, notably in the distal portions of the extremities (255,257). Pains, cramps, and paresthesias are common.
In the experience of Harding, reported before the discovery of frataxin, deep tendon reflexes were absent in 74% of patients, with the patellar reflex being lost in 99%. An extensor plantar response could be elicited in 89% (257,258). In the series of Pandolfo, controlled by determination of genotype, areflexia in the lower extremities was seen in 75% of cases and an extensor plantar response was seen in only 62% of cases (260).
MRI studies can show enlargement of the fourth ventricle and atrophy of the superior vermis, brainstem, and spinal cord (259). The somatosensory-evoked potentials recorded over the clavicles are abnormal even in the earliest stages of the disease (263). They are unaccompanied, however, by a delay in peripheral nerve conduction. Sphincter disturbance is rare except for occasional urgency of micturition (257). Neither mental retardation nor dementia was encountered in Harding’s large series (257,258). Electrocardiographic and echocardiographic evidence of myocarditis has been found in 80% to 90% of patients (259,260). T-wave abnormalities and congenital heart block are particularly common (264). Echocardiographic abnormalities are seen early; most commonly interventricular septal hypertrophy is seen (257). About 10% of patients develop diabetes mellitus and about 20% have carbohydrate intolerance (260).
The patient with an advanced case of FRDA is bedridden with dysphagia and other bulbar signs. Death is caused by inanition or, more commonly, myocarditis with intractable congestive failure.
Heterozygotes for FRDA are neurologically normal. According to Harding, they demonstrate neither pes cavus nor scoliosis (257).
When patients with clinically typical FRDA are examined for GAA trinucleotide expansion, a small proportion do not show an expanded allele. It is possible that these patients have point mutations or deletions in the gene for FRDA on both chromosomes (265) or have a genetically different disease with an FRDA phenotype. Conversely, triplet expansions have been seen in a number of patients with atypical ataxia and in patients presenting with generalized chorea (266).
Diagnosis
In the typical patient, the clinical diagnosis is suggested by the presence of childhood-onset progressive ataxia, skeletal deformities, an abnormal visual-evoked potential, and an abnormal echocardiogram result. The diagnosis is confirmed by determination of the size of GAA repeats, which can now be performed commercially. Ataxia-telangiectasia (see Chapter 12) is the second-most-common cause of childhood-onset progressive ataxia. It is distinguished clinically by cutaneous telangiectases, a history of frequent serious respiratory infections, absent or marked reduction of immunoglobulin A (IgA) globulins, an elevated α-fetoprotein, and a lack of skeletal deformities and sensory signs.
Harding described an autosomal recessive form of cerebellar ataxia in which deep tendon reflexes are preserved and in which optic atrophy, diabetes, and cardiac abnormalities cannot be demonstrated (257). Symptoms appear between 18 months and 20 years of age, and the progression of the illness is slower than in the classic form of FRDA. Most of these patients are now known to have the same mutation as is seen in typical FRDA (244).
Several other forms of ataxia having their onset in childhood and having an autosomal recessive inheritances have been recognized. They are summarized in Table 3.5.
An autosomal recessive infantile-onset spinocerebellar ataxia (IOSCA) is seen not infrequently in Finland and is probably identical with an entity once termed Behr disease. Symptoms develop during the second year of life and are highlighted by cerebellar ataxia, a peripheral sensory neuropathy, deafness, optic atrophy, athetosis, and seizures. Hypogonadism can be documented in female patients (269,270). Pathologic examination shows atrophy in the spinal cord: the dorsal roots, posterior columns, and posterior spinocerebellar tracts. There is a severe neuronal loss in the dorsal nucleus. The dentate nuclei and inferior olives are also atrophic (279).The gene for this condition has been mapped to chromosome 10q24 (280).
A recessively inherited ataxia caused by a mutation of the α-tocopherol transfer protein has a clinical picture that resembles FRDA (268). Age of onset is younger than 20 years in patients who stem from the Mediterranean region but is later in Japanese cases.
The other recessively inherited ataxias are even rarer. These include an entity characterized by cerebellar ataxia, mental deficiency, congenital cataracts, impaired physical growth, and various skeletal anomalies. It was first described by Marinesco and Sjögren, Garland, and others (275,276). The brain shows marked cerebellar atrophy, especially affecting the vermis, with almost complete loss of Purkinje and granule cells and with gliosis of the dentate
nucleus. The presence of a myopathy is suggested by EMG and by the presence of fiber-type disproportion, fibrosis, and lipid deposition on biopsy (276). Electron microscopic studies on sural nerve biopsies demonstrate numerous abnormally enlarged lysosomes. Lysosomal enzyme study results, however, have been normal. The condition has been mapped to chromosome 5q31 (281).
nucleus. The presence of a myopathy is suggested by EMG and by the presence of fiber-type disproportion, fibrosis, and lipid deposition on biopsy (276). Electron microscopic studies on sural nerve biopsies demonstrate numerous abnormally enlarged lysosomes. Lysosomal enzyme study results, however, have been normal. The condition has been mapped to chromosome 5q31 (281).
TABLE 3.5 Recessively Inherited Ataxias | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
A recessively inherited form of spastic ataxia has been described in an isolated French Canadian community of Quebec (Charlevoix-Saguenay syndrome) (267). The condition has been mapped to chromosome 13q11, and the gene (SACS) codes for a protein that is believed to be involved in chaperone-mediated protein folding (282).
Treatment
Children with FRDA should remain active for as long as possible and participate in physical therapy and programmed remedial exercises. These should focus on balance training and muscle strengthening. In my experience, the cardiomyopathy does not interfere with such an exercise program. Orthopedic surgery for skeletal deformities, particularly progressive scoliosis, which is a major cause for morbidity, might be necessary if bracing does not arrest the progression of the scoliosis (283). The antioxidant idebenone, a synthetic analogue of coenzyme Q10, has induced a significant reduction of cardiac hypertrophy at doses as low as 5 mg/kg and improved cardiac function, but in the experience of Buyse and coleagues did not arrest the progression of ataxia (284). The effectiveness of much higher doses (60 to 70 mg/kg) on the neurologic manifestations is under trial (285).The use of mitochondrially targeted antioxidants (mitoquinone–ubiquinone conjugated to a lipophilic cation that enchances its mitochondrial uptake) is also under investigation. Nontargeted antioxidants such as coenzyme Q10 stabilize or reverse the cardiomyopathy but at the doses used have no effect on the ataxia (285). In the experience of the Ataxia Clinic at the University of California, Los Angeles, patients feel better when their carbohydrate intake is restricted. Children with FRDA should be seen by cardiologists and orthopedic surgeons at regular intervals (once or twice a year). With good supportive care, patients can expect to live into their 40s or 50s.
Dominant Spinocerebellar Ataxias (SCAs) (Olivopontocerebellar Atrophies)
The SCAs represent some 25 genetically distinct autosomal dominant conditions, some of which were in the past designated as olivopontocerebellar atrophies and whose prototypes were first described by Menzel in 1890, Déjérine and Thomas in 1900, and Holmes in 1907 (286,287,288). Clinically, they manifest by progressive cerebellar ataxia, tremor, speech impairment, and, in some instances, marked extrapyramidal signs, cranial nerve palsies, and peripheral neuropathy (see Table 3.6). Approximately one-third of cases with autosomal dominant ataxia do not correspond to any of the mapped conditions, so that, over time, further genetic subtypes will undoubtedly be delineated.
SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17 are caused by the inheritance of an expansion of CAG repeats coding for an extended polyglutamine tract. This expansion is located in the coding region of the respective genes. The genes SCA8, SCA10, and SCA12 have expansions of CTG, ATTCT, and CAG repeats, respectively, in their noncoding region. Although the mean age of onset of these disorders is during the fourth decade of life, the conditions have sometimes been encountered in children. The length
of the expanded repeat tends to increase from generation to generation, and there is an inverse correlation between the size of the repeat and the age when symptoms become apparent. As a result, a progressively earlier onset of symptoms occurs in successive generations, a phenomenon termed anticipation.
of the expanded repeat tends to increase from generation to generation, and there is an inverse correlation between the size of the repeat and the age when symptoms become apparent. As a result, a progressively earlier onset of symptoms occurs in successive generations, a phenomenon termed anticipation.
TABLE 3.6 Dominantly Inherited Ataxias | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
SCA1
In the series of Benton and colleagues, some 6% of families with autosomal dominant SCA have a defect in SCA1 (310). In the Italian series of Brusco and coworkers, SCA1 accounted for 21% of patients with hereditary ataxia (310a). SCA1 codes for a protein termed ataxin 1. It is found ubiquitously and is a nuclear protein in all cells with the exception of the cerebellar Purkinje cells, where it is cytoplasmic as well as nuclear. As is the case for HD, the extended polyglutamine tract is believed to induce neuronal degeneration in an as-yet-undefined manner. Lin and coworkers attributed the toxic effects of the expanded polyglutamine tracts to downregulation of a number of neuronal genes (311).
Expanded repeats tend to increase in length more with paternal than with maternal transmission; as a consequence, the majority of patients with juvenile onset are offspring of affected men. In the series of Pandolfo and Montermini, 16% of SCA1 patients developed symptoms before 15 years of age (289). Characteristic symptoms include ataxia, ophthalmoplegia, and pyramidal and extrapyramidal signs. Neuropathologically, patients with SCA1 show gross atrophy of the cerebellum, cerebellar peduncles, and basis ponti. The Purkinje cells appear most affected. A marked reduction in the number of granule cells and neuronal loss in the dentate nucleus are seen. Pathologic changes can also involve the basal ganglia, spinal cord, retina, and peripheral nervous system.
SCA2
SCA2 is phenotypically similar to SCA1, but movement disorders and a neuropathy are common. The gene for the disease codes for a highly basic protein, ataxin 2, having no similarity with any other proteins. Ataxin 2 is found in all parts of the brain, but an ataxin 2–binding protein is
predominantly expressed in the cerebellum (312). In the series of Benton and colleagues, SCA2 was encountered in 21% of families with autosomal dominant cerebellar ataxia (310). In the Italian series of Busco and coworkers, SCA2 accounted for 24% of cases of dominant cerebellar ataxia (310a). SCA2 is the most common dominant cerebellar ataxia in India. SCA2 is also the most common dominant cerebellar ataxia in children seen in the Ataxia Clinic at the University of California, Los Angeles (S. Perlman, personal communication, 2004). The onset of neurologic symptoms is before 10 years of age in some 3% of patients. In these children, the disease is nearly always paternally transmitted.
predominantly expressed in the cerebellum (312). In the series of Benton and colleagues, SCA2 was encountered in 21% of families with autosomal dominant cerebellar ataxia (310). In the Italian series of Busco and coworkers, SCA2 accounted for 24% of cases of dominant cerebellar ataxia (310a). SCA2 is the most common dominant cerebellar ataxia in India. SCA2 is also the most common dominant cerebellar ataxia in children seen in the Ataxia Clinic at the University of California, Los Angeles (S. Perlman, personal communication, 2004). The onset of neurologic symptoms is before 10 years of age in some 3% of patients. In these children, the disease is nearly always paternally transmitted.
SCA3
SCA3, also known as Machado-Joseph disease, is also phenotypically similar. In the series of Pandolfo and Montermini, the onset of neurologic symptoms was before 15 years of age in 7% of patients (289). In Germany, this condition is the most common of the autosomal dominant cerebellar ataxias, being found in 42% of the families with autosomal dominant ataxia (313). In the series of Benton and colleagues, this condition was seen in only 8% of families (310). SCA3 accounted for only 1% of cases in the Italian series of Brusco and coworkers (310a). The mechanism of neuronal damage in this condition is not yet clarified. Schmidt and coworkers found the polyglutamine-expanded protein displayed an increased propensity to misfold and aggregate and that the nuclear aggregates of the mutant protein form the ubiquinated neuronal intranuclear inclusions seen in the pons and substantia nigra (314).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


