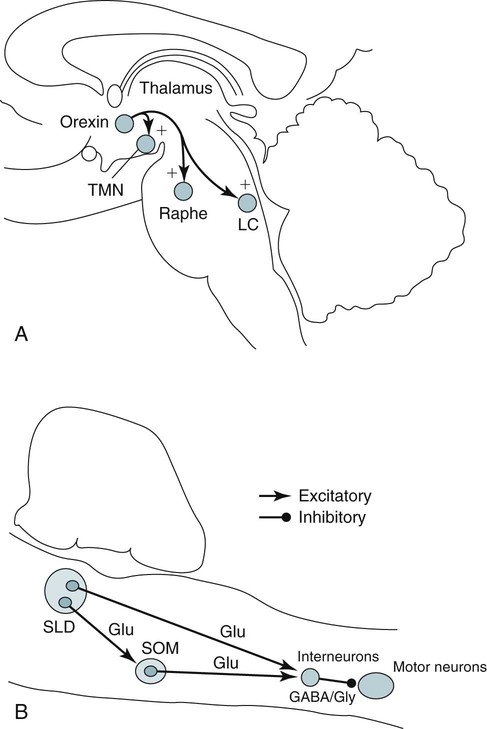
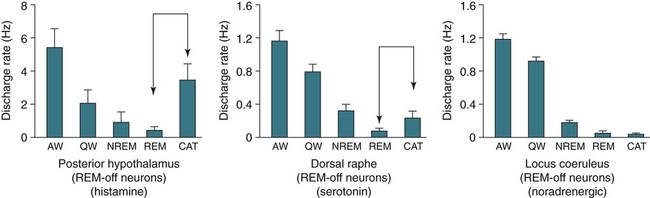
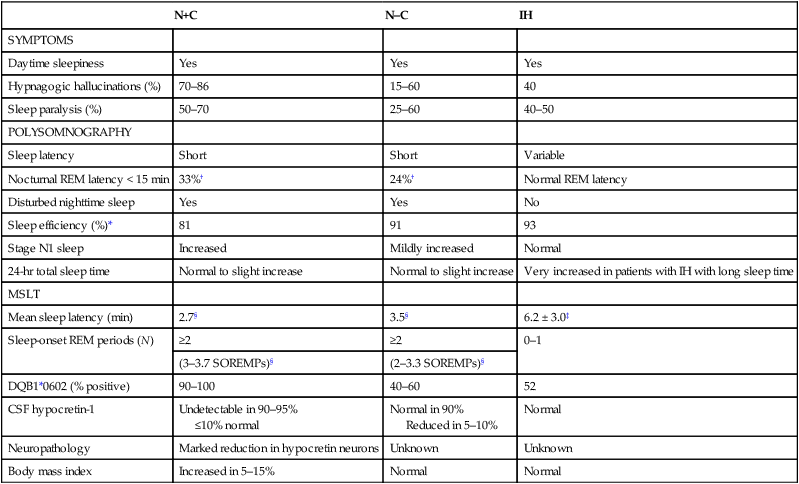
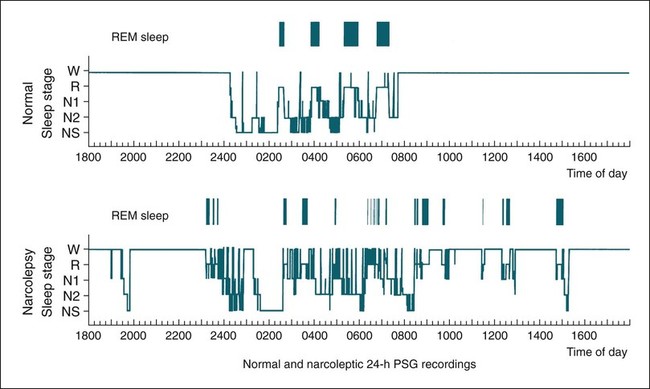
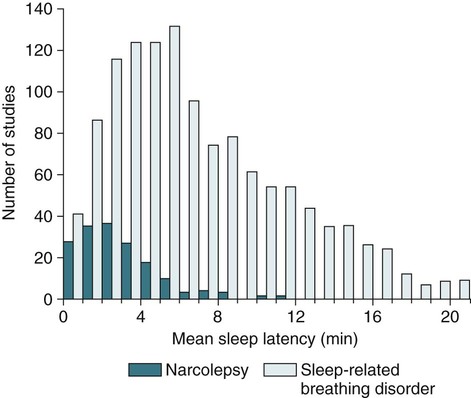
• N+C and N–C are two disorders characterized by EDS and symptoms related to the abnormal regulation of wake and sleep. A short REM latency and intrusion of REM sleep features into wake are characteristic of the disorders. • Cataplexy is characterized by temporary muscle weakness induced by emotional stimuli. The affected individual is conscious and the duration of weakness is usually seconds to minutes. Hearing or telling a joke and laughter are the most common triggers. • Approximately 60–70% of patients with narcolepsy have cataplexy. • Cataplexy is the only symptom virtually specific for narcolepsy. Cataplexy in the absence of narcolepsy occurs only in patients with a few rare genetic disorders. Patients with these disorders have abnormal neurological function. • Approximately 90–95% of patients with N+C have low or absent Hcrt1 in the CSF and absent Hcrt-producing neurons in the lateral hypothalamus. • Other manifestations of narcolepsy include sleep paralysis, sleep-related hallucination, automatic behavior, disturbed sleep, and the RBD. • The hallucinations of narcolepsy that occur at nocturnal sleep onset are called hypnagogic hallucinations and those on awakening are termed hypnopompic hallucinations. • For N+C, the MSLT is confirmatory. The diagnosis of N–C depends on demonstration of sleep-onset REM periods in the absence of other sleep disorders (such as sleep apnea) that can cause a short REM latency. • The MSLT criteria for narcolepsy are a mean sleep latency ≤ 8 min and two or more sleep-onset REM periods. • IH with a long sleep time in contrast to IH without a long sleep time is characterized by a typical sleep duration of longer than 10 hours. The diagnosis depends on exclusion of other disorders that could explain the EDS. • The MSLT criteria for IH are mean sleep latency <8 minutes and fewer than 2 sleep REM-onset periods. • Treatments for the EDS of narcolepsy include modafinil (and armodafinil), stimulants (methylphenidate and others), and sodium oxybate. • Treatments for cataplexy include sodium oxybate (FDA approved) and non–FDA-approved medications including TCAs, SSRIs, and SNRIs. These medications also decrease sleep paralysis and HHs. • Treatments for the EDS of IH include modafinil (armodafinil) and stimulants. The hypersomnias of central origin are those conditions in which hypersomnia is due to dysfunction of the central nervous system (CNS). The dysfunction is not due to sleep-related breathing disorders (SRBDs), circadian rhythm sleep disorders, or other causes of disturbed nocturnal sleep. Although the nocturnal sleep in some of these disorders is not normal, the daytime sleepiness is out of proportion to the degree of sleep disturbance. The International Classification of Sleep Disorders, 2nd edition (ICSD-2)1 lists a number of disorders in the category “Hypersomnias of Central Origin” (Box 24–1). This chapter discusses the disorders with the emphasis on narcolepsy with and without cataplexy. Narcolepsy is a chronic disorder characterized by excessive daytime sleepiness (EDS) and symptoms related to the abnormal regulation of wakefulness and sleep. It is characterized by a short rapid eye movement (REM) latency and intrusion of REM sleep features into the waking state. Approximately 60% to 70% of all patients with narcolepsy have cataplexy.1–4 Cataplexy consists of temporary muscle weakness following emotional stimuli. Narcolepsy with cataplexy (N+C) and narcolepsy without cataplexy (N–C) are now classified as two distinct disorders but share many of the same features1–4 (Table 24–1). TABLE 24–1 As the name implies, definitive cataplexy is present in patients with N+C. Following the discovery of hypocretin (Orexin) peptides in 1998,5,6 it was learned that the majority of patients with N+C have low or undetectable levels of hypocretin-1 (Hcrt1) in the cerebrospinal fluid (CSF).7 However, much more much remains to be determined about the pathophysiology of narcolepsy. In the absence of cataplexy, a diagnosis of N–C depends on demonstration of sleep-onset REM (very short REM latency) and an absence of other disorders to explain this finding.1–4 The key symptoms of narcolepsy were described by Westphal in 1877.8 The term narcolepsy, meaning “to seize with drowsiness,” was coined by Gelinau.8 Aidie termed the loss of muscle tone as “cataplexy” in the early 20th century.9 Yoss and Daly10 described the classic tetrad of daytime sleepiness, cataplexy, hypnagogic hallucinations, and sleep paralysis (SP). In 1960, Vogel11 reported that sleep-onset REM periods were associated with narcolepsy. Initially, the association of narcolepsy with the human leukocyte antigen (HLA) DR2 antigen was described in Japanese patients.12 Later, the HLA DQB1*0602 was found to be strongly associated with narcolepsy in white and African American populations.13 In 1998, two groups simultaneously reported the existence of hypocretin (Orexin) peptides present in the lateral and posterior hypothalamus.5,6 Cleavage of a single precursor protein produces the peptides hypocretin-1 (Orexin A) and hypocretin-2 (Orexin B). In 2000, the association of narcolepsy with cataplexy and low or undetectable levels of CSF Hcrt1 was described.7 Narcolepsy is present in about 1/2000 persons. Approximately 60% to 70% of patients with narcolepsy have cataplexy.14 Men and women are affected equally. The average age of onset in most studies is between 20 and 30 years. However, narcolepsy can begin at any age. Dauvilliers and coworkers15 found two peaks in the age of onset with one around age 15 and the other around 35 years of age. Familial canine narcolepsy is transmitted as a single autosomal recessive gene (canarc-1) with complete penetrance.16 This form of narcolepsy is due to an abnormal Hcrt2 receptor. However, the human form of the narcolepsy is not a simple genetic disease. Although the majority of cases of human narcolepsy are sporadic, there have been numerous reports of familial narcolepsy in the literature.16,17 Studies of families of patients with narcolepsy revealed that the risk of a first-degree relative of a narcoleptic developing N+C is 1% to 2%, a 10 to 40 times higher risk than in the general population.16 However, studies of identical twins show a high degree of discordance for N+C. That is, if one twin has narcolepsy, the other twin will have or develop narcolepsy only about 25% to 31% of the time16 (Table 24–2). Thus, factors other than genetics are important for the development of human narcolepsy. TABLE 24–2 N+C is strongly linked to specific HLAs. Approximately 90% to 95% of patients with N+C have the DQB1*0602 allele regardless of race16 (see Table 24–2). However, this allele is present in about 12% of Japanese, 25% of whites, and 38% of African Americans without the syndrome. The percentage of patients with N–C that are DQB1*0602 positive is lower (40–60%). In general, patients with narcolepsy who are DQB1*602-positive have more severe symptoms.2 Hcrt neurons located in the lateral or posterior hypothalamus project widely in the CNS (Fig. 24–1A). They augment the activity of brain areas active during wakefulness (tuberomammillary nucleus [TMN]—histamine, locus coeruleus—norepinephrine [NE], and dorsal raphe—serotonin).18–23 Their activity is believed to stabilize the sleep-wake system to prevent abrupt transitions between sleep and wakefulness. Histaminergic neurons are located exclusively in the TMN of the posterior hypothalamus and project to various brain regions associated with regulation of sleep-wake cycles. Activity of these histaminergic neurons is believed to help promote wakefulness. Hcrt neurons project to the TMN and stimulate the TMN neurons via Hcrt2 receptors. As noted previously, canine N+C is usually secondary to a defect in the receptor for Hcrt2. Human N+C is associated with absent or very low CSF levels of Hcrt1 (90–100% of patients).15,24–26 Brains of patients with N+C have reduced staining for Hcrt in the hypothalamus.27,28 N+C is associated with a loss of approximately 90% of Hcrt neurons. Melanin-concentrating hormone neurons, which are intermixed with Hcrt cells in the normal brain, were not reduced in number, indicating that cell loss is relatively specific for Hcrt neurons. It is believed that hypothalamic Hcrt cells are destroyed before disease onset. Mutations in Hcrt genes do not appear to be the cause of most human narcolepsy.29 Of note, up to 10% of patients with N+C have normal CSF Hcrt1 levels.25,26 Thus, other abnormalities of the Hcrt system or other mechanisms must be the cause of the syndrome in these patients. The cause of the loss of Hcrt cells in patients with N+C is unknown. Because of the association of narcolepsy with HLA antigens, an autoimmune mechanism has been hypothesized but has been difficult to document.30 Elevated antistreptococcal antibodies were found in one study of patients with recent narcolepsy onset.31 Another study found that narcolepsy is also associated with a polymorphism in the T-cell receptor alpha gene.32 Recently, several studies have found that some patients with narcolepsy have elevated levels of antibodies against a protein known as Tribbles homolog 2 (TRIB2).33–35 In one study, antibodies were found in about 25% of patients with N+C but in only 3.5% in patients with N–C or 4.5% in normal controls.35 The presence of TRIB2 antibodies was associated with a short duration since disease onset (recent onset of narcolepsy). TRIB2 is produced in Hcrt neurons, providing some of the firmest evidence yet for an autoimmune cause of narcolepsy. However, whether the Tribbles antibodies cause Hcrt damage, occur secondary to damage of the Hcrt neurons, or are simply an associated phenomenon secondary to another cause of Hcrt neuron injury is unknown.36 Two investigations have documented low CSF histamine in patients with narcolepsy (with and without cataplexy) and idiopathic hypersomnia (IH).37,38 As noted previously, histamine secretion is believed important for maintaining wakefulness. It is not clear whether low CSF histamine is a cause or an effect of EDS in these patients. Recall that Hcrt neurons stimulate histaminergic neurons in the TMN. However, CSF histamine was low even in patients without Hcrt ligand deficiency (N–C and IH). Therefore, other defects beside Hcrt deficiency must be involved in producing low CSF histamine levels. The pathophysiology of N–C is not well understood. A small percentage (5–10%) of N–C patients have a reduced CSF Hcrt (most are positive for HLA DQB1*0602). N–C patients negative for the HLA DQB1*0602 almost always have normal CSF Hcrt1 levels.7,25,26 The etiology of N–C is currently unknown but may represent a dysfunction of the Hcrt system without total loss of Hcrt-producing cells. Thannickal and coworkers39 examined the brain of one patient with N–C and found a 33% reduction in hypocretin cells (compared to normals) with maximal loss in the posterior hypothalamus. Thus, partial loss of Hcrt neurons and other alterations in the Hcrt system without a reduction in CSF Hcrt could be the etiology of N–C. As noted previously, CSF histamine is low in N–C patients.37,38 This may be a marker of hypersomnia, or reduced histamine secretion could be part of the pathogenesis of the disorder. The mechanisms inducing cataplexy are still under investigation.40–42 The atonia of REM sleep is thought to be due to inhibition of the spinal alpha motor neurons of the anterior horn cells by glycine and gamma-aminobutyric acid (GABA) secreted by spinal interneurons (see Fig. 24–1B). The interneurons are activated directly by projections from areas in the pons responsible for atonia or via an intermediate relay area in the ventral medulla. Glutamate is believed to activate the spinal interneurons. The atonia neurons of the pontine reticular formation are located ventral to the locus coeruleus and are often called the subcoeruleus (SubC) or sublaterodorsal nucleus (SLD). The neurons are believed to be glutaminergic. During REM sleep, there also appears to be inhibition of pathways normally promoting alpha motor neuron activity. Cataplexy and the atonia of REM sleep may share some common pathways, but there are important differences. John and colleagues41 reported that histaminergic neurons in the ventral posterior lateral hypothalamus remain active during cataplexy as opposed to REM sleep (Fig. 24–2). Because histamine is associated with wakefulness, this may at least partially explain why patients remain conscious during cataplexy but not REM sleep. The other important difference between REM-associated atonia and cataplexy is the triggering emotional stimulus for cataplexy. Neural pathways involving the amygdala are thought to be important for the emotional induction of cataplexy. Similarities and differences between N+C, N–C, and IH are outlined in Table 24-3. IH is discussed in detail in a later section. TABLE 24–3 Features of Narcolepsy Syndromes versus Idiopathic Hypersomnia *Sleep efficiency = total sleep time/total recording time. †From Aldrich MS, Chervin RD, Malow BA: Value of the multiple sleep latency test (MSLT) for the diagnosis of narcolepsy. Sleep 1997;20:620–629. ‡From Arand D, Bonnet M, Hurwitz T, et al: A review by the MSLT and MWT Task Force of the Standards of Practice Committee of the AASM. The clinical use of the MSLT and MWT. Sleep 2005;28:123–147. §From Scammel TE: The neurobiology, diagnosis, and treatment of narcolepsy. Ann Neurol 2003;53:154–166. Adapted from Scammel TE: The neurobiology, diagnosis, and treatment of narcolepsy. Ann Neurol 2003;53:154–166, with permission. The EDS of narcolepsy can occur as discrete “sleep attacks” or as constant sleepiness with intermittent worsening. Unrelenting EDS is usually the first and most prominent symptom of narcolepsy.1–4,43 Sleepiness may occur throughout the day, regardless of the amount or quality of prior nighttime sleep. Sleep episodes may occur at work and social events, while eating, talking, and driving, and in other similarly inappropriate situations. Napping is usually restorative but provides only temporary relief. It has been said that falling asleep while standing or eating is especially suggestive of narcolepsy. Even though patients may sleep at every hour of the day, the total sleep time over a 24-hour period is normal or only slightly increased.44,45 Rogers and associates44 performed 24-hour polysomnography (PSG) in a group of narcolepsy patients who were “well controlled.” Of 25 subjects with narcolepsy, 10 had no daytime sleep but the other 14 averaged 2.7 naps and 76.2 minutes of sleep during the day (compared with 4.8 min for controls).44 The two groups did not differ in the total amount of sleep or the total amount of REM sleep in 24 hours. Figure 24–3 compares the 24-hour hypnograms of a patient with narcolepsy and a normal subject. The hypnogram shows sleep episodes scattered throughout the day, and some of the daytime sleep episodes contain REM sleep. In a study comparing narcoleptics and normal subjects using 24-hour ambulatory monitoring, Broughton and coworkers45 found no increase in the total amount of sleep in 24 hours. However, narcoleptics had more daytime and less nighttime sleep than normal persons. The sleep-related hallucinations of narcolepsy that occur at nocturnal sleep onset are called hypnagogic hallucinations (HGHs) and those on awakening are termed hypnopompic hallucinations (HPHs). The hallucinations are typically bizarre and may be frightening. Patients sometimes have a fair degree of insight that they are hallucinatory in nature, but often consider them no less frightening. The duration is usually less than 10 minutes, and the frequency is quite variable. Visual imagery is the predominant feature for many patients, sometimes with intense colors. A commonly described vision is that of an animal or stranger in the room. Auditory or vestibular hallucinations (sensation of falling) may also occur. The percentage of narcolepsy patients reporting sleep-related hallucinations varies from 20% to 70% depending on whether data are obtained from questionnaire or physician interview.2–4 A questionnaire study by Aldrich4 found the proportion of N+C patients reporting sleep-related hallucinations was higher than in the N–C or IH groups. Sleep-related hallucinations (especially HPH) can be associated with SP (discussed in the next section) and can also occur in normal individuals. SP is partial or complete paralysis during the onset of sleep or upon awakening. Patients are awake and conscious during the attack. There is no emotional precipitant. The episodes may last longer than a typical cataplectic attack. People who experience SP sometimes experience hallucinations simultaneously. Studies have found that up to 50% to 80% of patients with N+C report SP, with a lower percentage in the N–C group.4,43 Although HH/HPH, SP, and daytime sleepiness are the classic narcolepsy symptoms, patients often report other associated symptoms that can be significant (see Table 24–1). Narcolepsy patients often experience disturbed nighttime sleep with tossing and turning in bed, leg jerks, nightmares, and frequent awakenings. The amount of stage N1 sleep is increased. In general, N+C patients have more disturbed sleep than N–C patients.1–4 Automatic behavior is present in up to 50% of patients. Automatic behavior is defined as performing a seemingly purposeful task with no clear memory of having performed the activity. For example, patients report driving a car and not remembering the trip. They may find themselves doing activities that make no sense like putting salt in iced tea. These episodes typically involve activities that are habitual or not demanding of skill. Inattentiveness related to drowsiness may occur. Aldrich4 reported that the proportions of patients reporting automatic behavior were similar in groups of N+C and N–C patients. Patients with narcolepsy may also have the rapid eye movement sleep behavior disorder (RBD).46 In this disorder, skeletal muscle atonia is absent during REM sleep and dreams may be acted out. Because the Hcrt system has affects on appetite, it is not surprising that some patients with N+C have an increased body-mass index.4,47 Obstructive sleep apnea (OSA) is also not uncommon.48 If cataplexy is not present in a patient with OSA, narcolepsy may be suspected only if daytime sleepiness persists after adequate treatment of the sleep apnea. Periodic leg movements in sleep (PLMS) are also common in narcolepsy.49 PLMS may be present during REM sleep, a feature uncommon in most patients with PLMS. As the name implies, cataplexy is an essential criterion for the diagnosis of N+C. Cataplexy is the only symptom relatively “specific” to narcolepsy.1–4,50 Isolated cataplexy or cataplexy with sleepiness (secondary narcolepsy) can occur in a few rare neurologic disorders associated with mental retardation and obvious neurologic deficits. These are discussed in a later section. In a patient with daytime sleepiness and a normal neurologic examination, cataplexy is virtually diagnostic of narcolepsy. Cataplexy is characterized by the sudden, temporary loss of bilateral muscle tone with preserved consciousness triggered by strong emotions such as laughter, anger, or surprise.43,50 After cataplexy episodes, patients have total recall of the entire event. If the episodes last longer than a few minutes, the patient may transition into REM sleep and experience HHs. A systematic survey of symptoms of muscle weakness associated with emotion in a large group of patients with daytime sleepiness found that weakness during joking (telling or hearing a joke), laughter, and anger were the most specific for cataplexy associated with narcolepsy.43,50 Involvement of the legs also seemed to be more specific for narcolepsy. When loss of muscle strength during cataplexy is severe, almost all of the voluntary muscles in the body are affected, leading to complete collapse. The muscles of the eyes are not affected during cataplexy; individuals can move their eyes during a cataplectic episode. Diaphragmatic activity is also not impaired. In one study, the legs and knees were most frequently affected during cataplexy.50 In milder cases of cataplexy, the loss in muscle strength can be quite subtle, involving only a few muscle groups or occurring unilaterally. For example, ptosis, difficulty speaking, or partial neck muscle weakness may occur (head nodding). In some patients, partial attacks are more frequent than complete attacks. Loss of muscle function may not be evident, and the patient may experience only a vague feeling of weakness. Patients may fall to the ground, and injuries do occur. However, most people are able to find support at the onset of an attack. The attacks do start abruptly but usually take several seconds to reach their maximum intensity. Episodes of cataplexy usually last from seconds to minutes; rarely does an attack last longer than 2 minutes. Clinical signs during an attack are the loss of muscle tone and loss of deep tendon reflexes including a loss or decrease in the H reflex. During an attack of cataplexy, there are cardiovascular changes consisting of increased blood pressure and decreased heart rate. The phenomenon of virtually continuous attacks of cataplexy (status catapleticus) can occur after sudden withdrawal of medications that suppress cataplexy. Patients with daytime sleepiness are invariably questioned about SP, HH/HPHs, and the severity and nature of the daytime sleepiness. However, none of these historical elements is specific for narcolepsy and they can occur in sleep apnea and other causes of daytime sleepiness including IH4 (Table 24–4). A history of cataplexy is the most important symptom to elicit. In patients with EDS, unequivocal cataplexy, and a normal neurologic examination, a clinical diagnosis of narcolepsy can be made with some certainty. Even if cataplexy is present, most physicians would still seek objective confirmation with PSG followed by a multiple sleep latency test (MSLT).1 Daytime naps are often refreshing in patients with narcolepsy. In contrast, naps are not commonly refreshing in patients with IH. The nocturnal sleep study is used to rule out other significant sleep disorders (sleep apnea) that might explain daytime sleepiness. Nocturnal PSG often reveals a short sleep latency and impaired sleep quality with increased stage N1 sleep and decreased stage N3 sleep. The total sleep time may be reduced but the amount of REM sleep is usually normal.51,52 Patients with N+C on average have lower sleep efficiency, lower amounts of stage N3 sleep, more stage N1 sleep, and more awakenings than those with N–C.2,4 PLMS are common in patients with narcolepsy.49,52 Sleep-onset rapid eye movement sleep (SOREM) is defined as a REM latency less than 15 minutes. Aldrich and colleagues53 found that 33% of N+C, 24% of N–C, and 1% of SRBD patients had a SOREM on nocturnal PSG. Of interest, the finding of a SOREM period on the nocturnal PSG had a 98.5% specificity and 68% positive predictive value (true positives/total number of positives) for the diagnosis of narcolepsy. Therefore, although a minority of patients with narcolepsy have a nocturnal SOREMP (sleep-onset rapid eye movement period) on a given PSG, the finding is highly suggestive of narcolepsy. This is especially true if no other factor is present to explain the short REM latency such as depression, OSA, recent withdrawal of REM-suppressing medication, or sleep deprivation. The MSLT is the standard objective test for the assessment of sleepiness and the diagnosis of narcolepsy53–55 (Table 24–5; see also Table 24–4). Overnight PSG during the patient’s habitual sleep period should precede the MSLT. The purpose of the PSG is to exclude other causes of excessive daytime sleepiness such as OSA and periodic limb movement disorder (PLMD). A sleep diary or actigraphy should document a regular and sufficient amount of sleep for at least 7 days before the study. Medications that can alter sleepiness (stimulants) or REM sleep should be withdrawn at least 15 days before testing. REM-suppressing medications can alter the ability to detect REM sleep. However, acute withdrawal of REM-suppressing agents can cause a false-positive test. TABLE 24–5 Multiple Sleep Latency Test Findings in Patients Evaluated for Daytime Sleepiness *Diagnosis made in patients with N–C by repeat MSLT if initial test negative. Adapted from Aldrich MS, Chervin RD, Malow BA: Value of the multiple sleep latency test (MSLT) for the diagnosis of narcolepsy. Sleep 1997;20:620–629. The ICSD-2 criteria for the diagnosis of narcolepsy include a mean sleep latency of 8 minutes or less and two or more SOREMPs during five naps.1 Aldrich and colleagues53 found that the MSLT had improved sensitivity when a sleep latency of 8 minutes rather than 5 minutes was used as a criterion for sleep latency in patients with N–C. A meta-analysis found the mean sleep latency for narcolepsy patients to be 3.1 ± 2.9 minutes.54,55 Up to 30% of the general population have a mean sleep latency less than 8 minutes. However, the finding of two or more SOREMPs is much more specific for narcolepsy. Up to 2% of the normal population will have two SOREMPs, and in other studies, approximately 6% of patients with SRBDs will have a mean sleep latency less than 8 minutes with two SOREMPs on an MSLT.53,56 A few patients with narcolepsy will occasionally have a mean sleep latency above 10 minutes. However, the mean sleep latency is less than 5 minutes in the vast majority of patients with narcolepsy (Fig. 24–4). The mean sleep latency in patients with SRBDs tends to be higher than in narcolepsy, although overlap does occur. Unfortunately, the MSLT is not very sensitive or completely specific for the diagnosis of narcolepsy. Aldrich and colleagues53 reviewed MSLT findings in patients evaluated for daytime sleepiness (see Table 24–5). A diagnosis of narcolepsy in patients without cataplexy required a repeat MSLT in some cases. In patients with N+C, a single MSLT was positive only about 70% of the time. Hence, a negative MSLT does not rule out the diagnosis of narcolepsy. In patients ultimately believed to have only an SRBD, approximately 6% had a positive MSLT53,56 (see Table 24–5). Although a high percentage of patients with N+C are positive for the HLA DQB1*602 antigen, this test is not very useful for ruling in narcolepsy (for either N+C or N–C). The HLA DQB1*0602 is found in 12% to 38% of the general population. Therefore, a positive result does not rule in N+C. Conversely, a negative test for presence of DQB1*0602 is not useful because up to 10% of N+C patients and 40% to 60% of N–C patients are negative for the antigen.1,2,13,57 Hcrt1 levels of the CSF can be assayed at a few centers. In patients with N+C, over 90% have very low or undetectable Hcrt1 levels. In one study of patients with many neurologic diseases, only patients with N+C and a few patients with Guillain-Barré had undetectable Hcrt levels.58,59 Patients with a number of neurologic diseases had levels that were lower than normal but still detectable. As noted previously, patients with N–C usually have normal levels of CSF Hcrt1. However, about 10% of N–C patients have low CSF Hcrt1. Most of these N–C patients with low CSF Hcrt1 are positive for the HLA DBQ1*602.58–60 The ICSD-2 diagnostic criteria for N+C1 are listed in Box 24–2. EDS for at least 3 months is required along with cataplexy. When possible, the diagnosis should be confirmed by an MSLT (sleep latency ≤8 min and two or more SOREMPs). A low CSF Hcrt1 level is considered confirmatory even in the absence of an MSLT meeting diagnostic criteria. Other sleep disorders causing the abnormal MSLT should be excluded. Whereas only daytime sleepiness and cataplexy are required to make the diagnosis of N+C, other manifestations such as sleep-related hallucinations, SP, and automatic behavior are often present (see Table 24–1). Only 10% to 15% of patients have the classic symptom tetrad of daytime sleepiness, cataplexy, HGH/HPHs, and SP. Patients with N+C tend to have a higher frequency of SP, HHs, and more severe sleepiness than N–C patients. During PSG, sleep is more disturbed in N+C patients than in N–C patients. On the MSLT, N+C patients tend to have more SOREMPs. Although 90% of N+C patients are positive for DQBQ1*602, this is not useful diagnostically. The manifestations of N+C generally begin between the ages of 15 and 30 years.43 However, this form of narcolepsy can present in the pediatric age group or in patients older than 60 years. EDS alone or in combination with HHs and/or SP is the presenting symptom in approximately 90% of patients.43 Cataplexy may develop several years after symptoms begin. However, most patients with N+C develop cataplexy within 3 to 5 years of the onset of daytime sleepiness.14,43 Rarely, cataplexy can precede daytime sleepiness. Typical findings include a short sleep latency and approximately 20% to 30% have a short nocturnal REM latency.53 Of interest, in one study, the presence of sleep-onset REM during the nocturnal sleep study had a high positive predictive value for the presence of narcolepsy.53 There tends to be an increase in stage N1 sleep. The ICSD-2 diagnostic criteria for N–C are listed in Box 24–3. Daytime sleepiness must be present for at least 3 months but typical cataplexy is NOT present. An MSLT shows a mean sleep latency of 8 minutes or less and two or more SOREMPs. The other manifestations of narcolepsy including SP, HHs, and automatic behavior may or may not be present. Compared with patients with N+C, a lower percentage of patients with N–C report SP or HH (see Table 24–3).2 The treatment of narcolepsy is usually broken into two components: (1) treatment of EDS and (2) treatment of cataplexy, SP, and HGH/HPHs.61–64 The treatment of daytime sleepiness includes conservative measures (adequate sleep time, good sleep hygiene, scheduled daily naps65), stimulant medications, alerting medications, and sodium oxybate (Table 24–6). Adequate control of daytime sleepiness can be attained in about 60% to 80% of patients. The stimulant medications used to treat the daytime sleepiness of narcolepsy include amphetamine (racemic), dextroamphetamine, methamphetamine, and methylphenidate. Dextroamphetamine and methylphenidate are U.S. Food and Drug Administration (FDA) approved for treatment of narcolepsy. A mixture of amphetamine and dextroamphetamine (~25% l-amphetamine and 75% d-amphetamine) is also available (Adderall). Some patients respond best to a mixture, although d-amphetamine may cause less anxiety. Methamphetamine is effective but has a high abuse potential. The FDA does not list narcolepsy as an indication for methamphetamine. The stimulant medications are indirect sympathomimetic stimulants (increase the synaptic availability of dopamine and norepinepherine). TABLE 24–6 Medications Used to Treat Excessive Daytime Sleepiness
Hypersomnias of Central Origin
Narcolepsy Syndromes
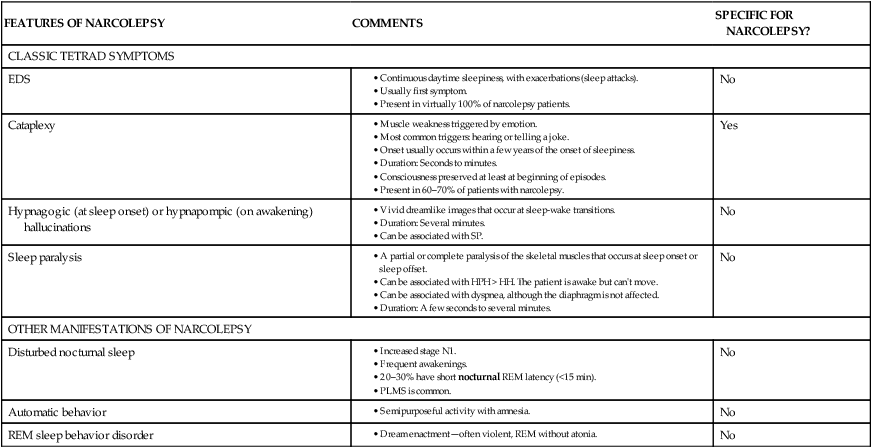
FEATURES OF NARCOLEPSY
COMMENTS
SPECIFIC FOR NARCOLEPSY?
CLASSIC TETRAD SYMPTOMS
EDS
No
Cataplexy
Yes
Hypnagogic (at sleep onset) or hypnapompic (on awakening) hallucinations
No
Sleep paralysis
No
OTHER MANIFESTATIONS OF NARCOLEPSY
Disturbed nocturnal sleep
No
Automatic behavior
No
REM sleep behavior disorder
No
History
Epidemiology
Genetics
INCIDENCE OF NARCOLEPSY
1/2000
FAMILIAL RELATIONSHIP
RISK FOR DEVELOPING NARCOLEPSY
Identical twin has narcolepsy with cataplexy.
25–31% of other twin has narcolepsy.
First-degree relative has narcolepsy.
1–2% (10–40 × higher risk).
HLA ANTIGEN PREVALENCE
HLA DQB1*0602 POSITIVE
General population.
12–38%.
N+C.
90% (≤10% negative).
N–C.
40–60%.
HYPOCRETIN
CSF HYPOCRETIN-1
N+C
Absent or low 90–100%.
Normal in up to 10%.
N–C
90% normal levels.
5–10% reduced levels (most + for DQB1*0602).
HLA Typing
Importance of Hcrt Neurons
Pathophysiology of Narcolepsy
N+C Group
N–C Group
Mechanisms of Cataplexy
Manifestations of Narcolepsy
N+C
N–C
IH
SYMPTOMS
Daytime sleepiness
Yes
Yes
Yes
Hypnagogic hallucinations (%)
70–86
15–60
40
Sleep paralysis (%)
50–70
25–60
40–50
POLYSOMNOGRAPHY
Sleep latency
Short
Short
Variable
Nocturnal REM latency < 15 min
33%†
24%†
Normal REM latency
Disturbed nighttime sleep
Yes
Yes
No
Sleep efficiency (%)*
81
91
93
Stage N1 sleep
Increased
Mildly increased
Normal
24-hr total sleep time
Normal to slight increase
Normal to slight increase
Very increased in patients with IH with long sleep time
MSLT
Mean sleep latency (min)
2.7§
3.5§
6.2 ± 3.0‡
Sleep-onset REM periods (N)
≥2
≥2
0–1
(3–3.7 SOREMPs)§
(2–3.3 SOREMPs)§
DQB1*0602 (% positive)
90–100
40–60
52
CSF hypocretin-1
Undetectable in 90–95%
≤10% normal
Normal in 90%
Reduced in 5–10%
Normal
Neuropathology
Marked reduction in hypocretin neurons
Unknown
Unknown
Body mass index
Increased in 5–15%
Normal
Normal
Excessive Daytime Sleepiness (N+C and N–C)
Sleep-Related Hallucinations (N+C and N–C)
Sleep Paralysis (N+C and N–C)
Disturbed Nocturnal Sleep and Other Symptoms: N+C, N–C
Cataplexy: N+C
Diagnostic Testing for Narcolepsy
History
Polysomnography
Multiple Sleep Latency Test
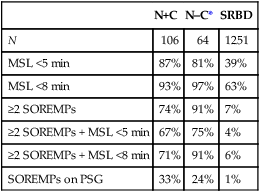
N+C
N–C*
SRBD
N
106
64
1251
MSL <5 min
87%
81%
39%
MSL <8 min
93%
97%
63%
≥2 SOREMPs
74%
91%
7%
≥2 SOREMPs + MSL <5 min
67%
75%
4%
≥2 SOREMPs + MSL <8 min
71%
91%
6%
SOREMPs on PSG
33%
24%
1%
HLA Typing
CSF Hcrt Levels
Diagnostic Criteria and Important Findings for N+C and N–C Forms of Narcolepsy
Narcolepsy with Cataplexy (N+C)
N+C Manifestations
Onset of N+C
Polysomnography
Narcolepsy without Cataplexy (N–C)
Treatment of Narcolepsy
Treatment of EDS
Medication (FDA PREGNANCY CATEGORY)
BRAND NAME
DOSE
MAXIMUM DOSE (DAILY)
HALF-LIFE (hr)
SELECTED SIDE EFFECTS
Methylphenidate*
(C)
Ritalin
10–30 mg bid or 10–20 mg tid (5, 10, 20 mg tabs)
100 mg
2–4
Nervousness, tremulousness, headache
Methylphenidate SR
Metadate and others
Concerta
10–20 mg SR qam (10, 20 mg tabs) + 10 to 20 mg short-acting in afternoon
18–36 mg SR qam (18, 27, 36, 54 mg tabs) + 10 to 20 mg short-acting in afternoon
Dextroamphetamine*
(C)
Dexedrine Dextrostat and others
5–60 mg qd
5–30 mg bid
(5, 10 mg tabs)
60 mg
10–30
Nervousness, tremulousness, headache
Amphetamine/dextroamphetamine
Adderall
(5, 7.5, 12.5, 15 mg tabs)
Dextroamphetamine
SR
10 mg SR qam
5, 10, 15 mg tabs
+10 to 20 mg short-acting in afternoon
SR
Amphetamine/dextroamphetamine XR
Adderall XR
5, 10, 15, 20, 25, 30 mg capsules
Methamphetamine* (C)
Desoxyn
5 to 60 mg qd (5, 10 mg tabs)
60 mg
12–34
Nervousness, tremulousness, headache
Lisdexamfetamine dimesylate*
(C)
Vyvanse
20, 30, 40, 50, 60 mg
70 mg
12–13
(duration of action)
Nervousness, tremulousness, headache
Modafinil†
(C)
Provigil
200 or 400 mg qd (100, 200 mg tabs)
400 mg
9–14
Headache, drug interactions, nervousness
Armodafinil†
(C)
Nuvigil
150–250 mg daily
150, 250 mg tabs
250 mg
10–14
Headache, drug interactions, nervousness
Sodium oxybate
(B)
Xyrem
4.5–9 g/night in 2 divided doses
9 g
1–3
Sedation, enuresis, respiratory suppression
Selegiline (C)
Eldepryl
20–40 mg
40 mg
9–14
Nausea, dizziness, confusion, dry mouth, requires low-tyramine diet, drug interactions
FDA approved for treatment of EDS in narcolepsy: modafinil, armodafinil, dextroamphetamine, methylphenidate, sodium oxybate.
Related posts:
 Obstructive Sleep Apnea Treatment Overview and Medical Treatments
Obstructive Sleep Apnea Treatment Overview and Medical Treatments
 Biocalibration, Artifacts, and Common Variants of Sleep
Biocalibration, Artifacts, and Common Variants of Sleep
 Oral Appliance and Surgical Treatment for Obstructive Sleep Apnea
Oral Appliance and Surgical Treatment for Obstructive Sleep Apnea
![]() Monitoring Respiration—Event Definitions and Examples
Monitoring Respiration—Event Definitions and Examples
 Central Sleep Apnea and Hypoventilation Syndromes
Central Sleep Apnea and Hypoventilation Syndromes
 Cardiac Monitoring during Polysomnography
Cardiac Monitoring during Polysomnography
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



