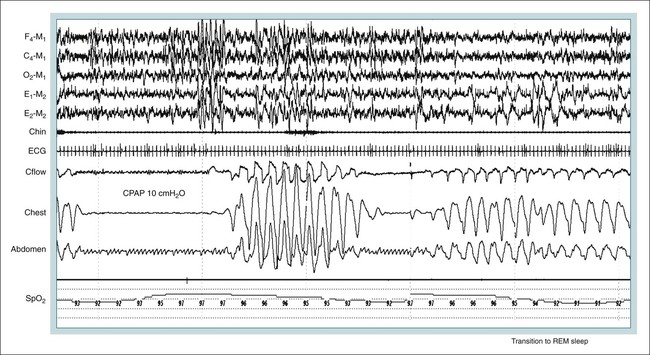
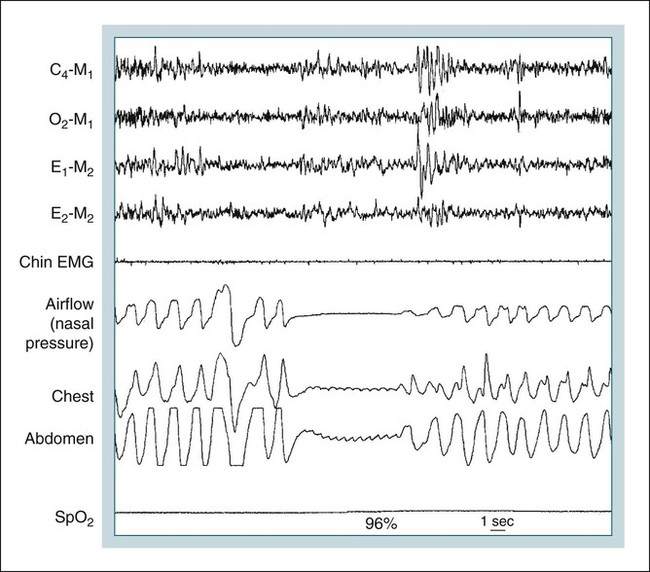
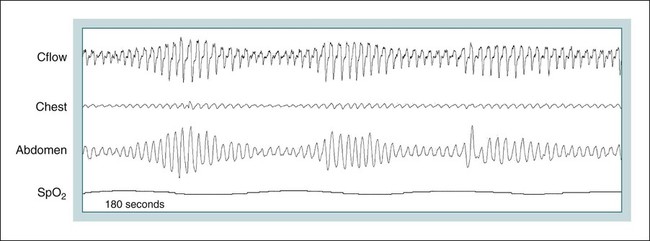
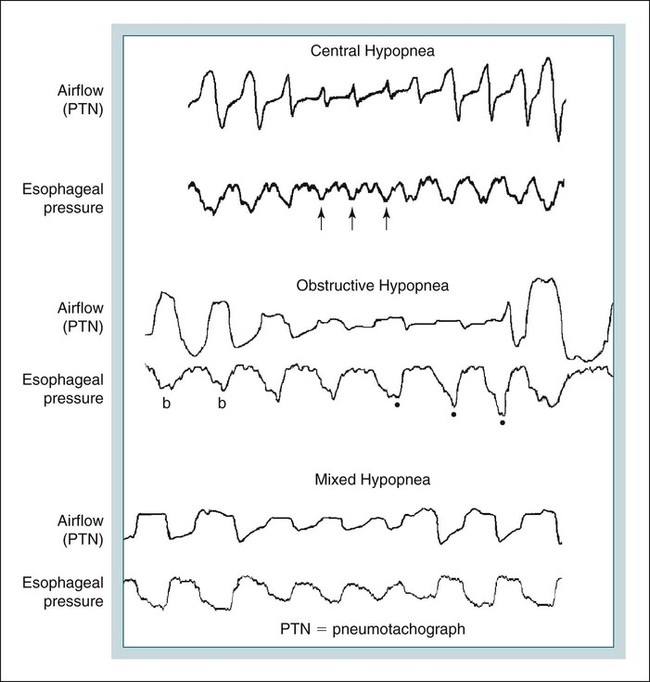
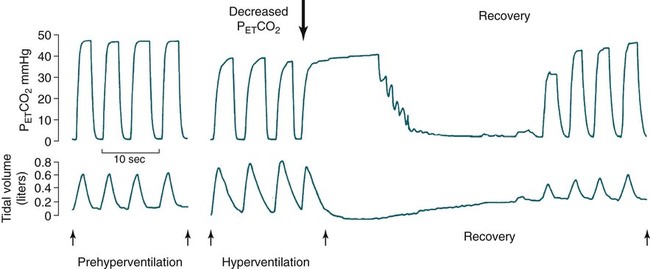
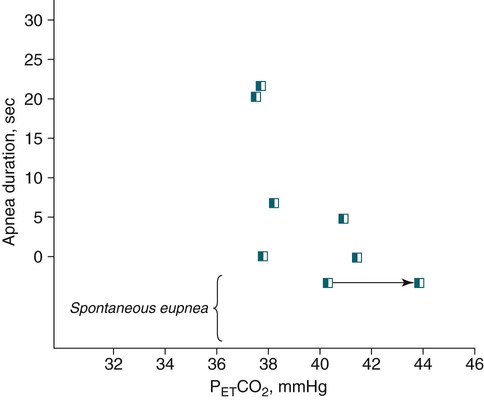
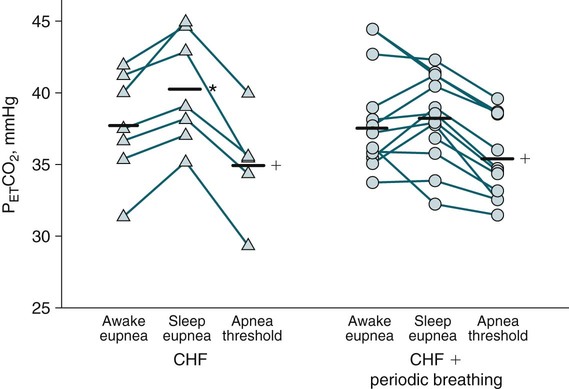
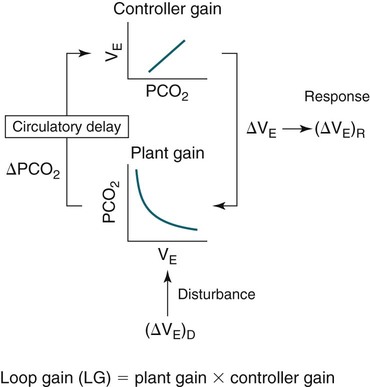
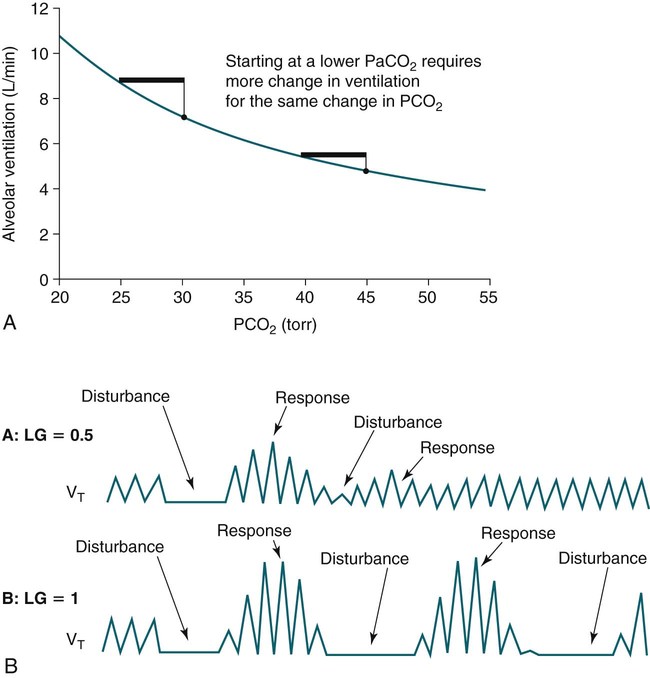
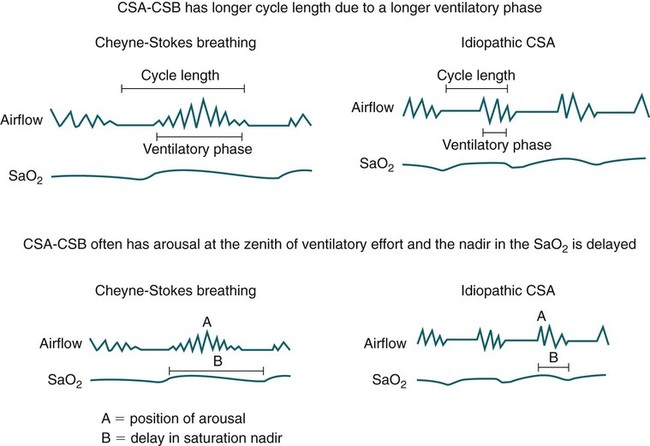
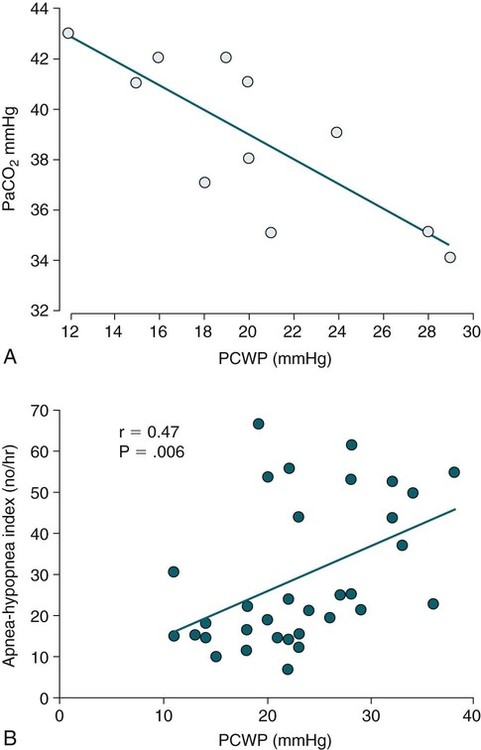
• Hypocapnic CSA occurs because the PaCO2 falls below the AT. The propensity for this type of central apnea to occur is increased when there is a small difference between the sleeping PaCO2 during sleep and the AT. • Hypocapnic CSA disorders include primary CSA (idiopathic CSA), CSB-CSA, HAPB, and some types of CompSA. • Hypocapnic CSA is often less severe during REM sleep because this sleep stage is associated with lower ventilatory responses to PaCO2 and PaO2, and ventilation varies with the phasic changes of this sleep stage. • CompSA is defined as the persistence or emergence of central apneas during a PAP titration in a patient with predominantly obstructive or mixed respiratory events during the diagnostic portion. • CompSA can occur in patients with a combination of OSA and CSB, patients taking opiates, or patients without an obvious etiology (idiopathic). • CompSA patients without CSB or opiate medication usually experience a resolution of CSA with chronic CPAP treatment. Patients with persistent CSA on CPAP require adaptive servo-ventilation or BPAP with a backup rate for effective treatment. • Hypercapnic CSA syndromes are characterized by sleep-related hypoventilation with variable amounts of CSA. Some patients may have a normal daytime PaCO2. • The CCHS is a rare disorder usually present from birth and is characterized by alveolar hypoventilation without evidence of lung, neuromuscular, or structural brainstem abnormalities. The disorder is due to mutations in the PHOX2b gene. • Opiate (narcotic)-associated sleep-disordered breathing can be manifested by long obstructive apneas, a slow respiratory rate, ataxic breathing, periodic breathing-central apneas or CompSA. Some patients have daytime hypoventilation. • Patients with RTCD or NMD may manifest daytime hypoventilation that worsens during sleep or exhibit hypoventilation only during sleep early in the disease course. Although classified under the hypercapnic CSA subgroup, many patients have relatively few discrete central apneas. • In RTCD/NMD patients, nocturnal gas exchange is usually the most abnormal during REM sleep. • Nocturnal oximetry may reveal significant desaturation even if the forced vital capacity is not severely reduced. If OSA is suspected PSG is indicated. • The treatment of choice for CSA/nocturnal hypoventilation in RTCD/NMD patients is NPPV. • In RTCD/NMD patients a daytime PaCO2 ≥ 45 mm Hg or nocturnal oximetry showing ≥ 5 minutes with an SaO2 ≤ 88% is considered an indication for nocturnal NPPV. • In patients with an NMD, an FVC < 50% of predicted or a maximal inspiratory force less than 60 cm H2O is also considered an indication for NPPV. • Patients with restrictive chest wall disorders may require relatively high pressure support. • Most clinicians would treat RTCD/NMD patients with BPAP using a backup rate (BPAP-ST). The central sleep apnea (CSA) syndromes include a diverse group of disorders associated with the presence of central apnea during sleep (Fig. 21–1).1–3 In some of the disorders discussed in this chapter, the patients have primarily nocturnal hypoventilation (increased arterial partial pressure of carbon dioxide [PaCO2]) due to inadequate tidal volume and/or respiratory rate with relatively few discrete central apneas. However, discussion of patients with central apnea and hypoventilation syndromes together is useful clinically because they have many similar aspects of pathophysiology and treatment. Central apnea in adults is defined as a cessation in airflow of 10 seconds or longer that is associated with an absence of respiratory effort.4 During diagnostic studies oronasal thermal sensor signal is used to detect apnea. During a positive airway pressure (PAP) titration the PAP flow signal is used. The diagnosis of a CSA syndrome requires that the majority of apneic events be central in nature. The exact proportion of central events required is not clear, with various authors diagnosing the CSA syndromes when 50% to 80% of the events are central. Traditionally, patients with CSA have accounted for less than 5% to 15% of patients with sleep apnea evaluated at most sleep centers. However, two factors have increased the number of CSA patients being evaluated. First, there has been an increased recognition of sleep-disordered breathing in congestive heart failure (CHF) and a substantial number of patients with systolic heart failure have Cheyne-Stokes breathing central sleep apnea (CSB-CSA) (Fig. 21–2). Second, recently, there has been more aggressive use of opiates to control pain. As discussed in this chapter, many patients develop central apnea as a result of the use of potent narcotics. Central apnea may occur either at baseline or once patients are exposed to PAP treatment. Thus, sleep centers can expect to see more patients with CSA. Many patients who exhibit central apneas also exhibit central hypopneas (see Fig. 21–2). The challenges in defining central hypopnea were discussed in earlier chapters. The American Academy of Sleep Medicine (AASM) scoring manual discourages identification of central hypopnea unless an accurate method to quantify respiratory effort is being used (esophageal pressure or respiratory inductance plethysmography).4 In general, central hypopneas are characterized by a proportionate decrease in both airflow and respiratory effort (Fig. 21–3).5 Usually, there is no snoring or chest-abdominal paradox and the nasal pressure or PAP device flow signal is fairly rounded (minimal or no signs of airflow limitation). There is a number of classifications of CSA and the hypoventilation syndrome but none of these classifications is entirely satisfactory.1,6 The International Classification of Sleep Disorders, 2nd edition (ICSD-2) lists five CSA syndromes (Box 21–1).6 Primary CSA is termed idiopathic central sleep apnea (ICSA) in this chapter, in keeping with much of the literature. Primary CSA, CSB-CSA, and high-altitude periodic breathing (HAPB) are hypocapnic forms of CSA. Patients with these syndromes have a normal or low PaCO2 during wakefulness. During sleep, these patients do not develop hypercapnia. In contrast, patients with CSA due to drug or substance and primary sleep apnea of infancy have normal or increased daytime PaCO2 and may develop or have worsening hypercapnia during sleep. The ICSD-2 lists five categories of hypoventilation syndromes (Box 21–2). Idiopathic nonobstructive hypoventilation is due to an abnormality of ventilatory control of unknown etiology. Congenital hypoventilation syndrome is due to abnormal ventilatory control due to a genetic abnormality. Three groups of disorders with hypoxemia or hypercapnia during sleep are listed. The first is due to lower airways obstruction (e.g., chronic obstructive pulmonary disease), the second is due to pulmonary parenchymal or vascular disorders (e.g., pulmonary fibrosis, interstitial lung disease), and the third is due to abnormality of the chest wall or neuromuscular disorders (e.g., kyphoscoliosis, amyotrophic lateral sclerosis [ALS], obesity hypoventilation syndrome [OHS]). Patients with the OHS are discussed in Chapter 15. The effects of chronic obstructive pulmonary disease on sleep (including hypercapnia) are discussed in Chapter 22. This chapter utilizes a classification of the CSA syndromes adapted from one proposed by Bradley and coworkers1 (Box 21–3) that subdivides patients into hypocapnic and hypercapnic groups. Patients with hypocapnic CSA tend to have low to normal daytime PaCO2 values. The disorders in this group include ICSA, CSB-CSA, HAPB, and treatment-persistent/-emergent CSA. The term complex sleep apnea (CompSA) has been used to identify patients who have primarily obstructive or mixed events during diagnostic studies but develop central apneas on PAP treatment (treatment-emergent central apneas) or have significant persistent central apneas on PAP treatment (treatment-persistent central sleep apnea).7 It is not unusual for patients with obstructive sleep apnea (OSA; predominantly obstructive apneas and hypopneas) to have some mixed or central apneas during diagnostic studies.8,9 PAP will eliminate central and mixed as well as obstructive apneas in many of these patients on the first treatment night (or during the initial PAP treatment).9 In other patients, central apneas will persist after airway obstruction is eliminated. In the early studies of the effect of treatment of OSA patients with tracheostomy (elimination of obstruction), patients were found to have residual central apneas that tended to resolve with chronic treatment.8 The term CompSA is also sometimes used to include patients with narcotic-associated sleep-disordered breathing who have persistent or emergent central apneas on PAP. Patients with this condition usually have high normal or mildly increased PaCO2 during wakefulness. In this chapter, this group is discussed under the hypercapnic CSA group. It is also not uncommon for patients with CHF to have a combination of obstructive or mixed apnea and CSB-CSA (Fig. 21–4).10 In some of these patients, application of PAP will eliminate obstruction but frequent central apneas of the Cheyne-Stokes type may persist or emerge. For this reason, the term CompSA is sometimes applied. These patients are discussed with the nonhypercapnic CSA disorders. Patients with CompSA without an obvious etiology (no narcotics or heart failure) are termed idiopathic CompSA for lack of a better terminology. These patients have an instability in ventilatory control either at baseline or due to PAP treatment. Because they have normal or low daytime PaCO2, they are discussed in the hypocapnic CSA sections. In normal individuals, there is a 2 to 8 mm Hg rise in PaCO2 during non–rapid eye movement (NREM) sleep. This is thought due to loss of the wakefulness drive, reduction of the hypercapnic and hypoxic ventilatory drives, and increased upper airway resistance (Table 21–1).11 The wakefulness drive is a poorly understood generalized augmentation of ventilation associated with the wakefulness state. During NREM sleep, ventilation is totally under metabolic control (chemoreceptors). Ventilatory control centers respond to information from the peripheral chemoreceptors including the carotid body (arterial partial pressure of oxygen [PaO2] and PaCO2) and medullary chemoreceptors (H+ due to changes in PaCO2).11 During rapid eye movement (REM) sleep, ventilation is irregular and nonmetabolic factors also affect ventilation. The hypercapnic and hypoxic ventilatory drives are lower during REM than during NREM sleep. In addition, during REM sleep, there is generalized skeletal muscle hypotonia.11–13 The contribution of accessory muscles of ventilation is either reduced or absent and ventilation depends entirely on the diaphragm. The loss of accessory inspiratory muscles can compromise the ability to maintain adequate ventilation, especially in patients with muscle weakness or a high work of breathing. During the phasic changes of REM sleep (associated with bursts of eye movements), there is often additional inhibition of upper airway muscles and diaphragmatic activity resulting in episodes of central apnea or reduced tidal volume (Fig. 21–5).12,13 Patients with hypercapnic CSA/hypoventilation syndromes usually have worse oxygenation and highest PaCO2 during REM sleep. It is of interest that some patients with hypocapnic CSA due to ventilatory control instability may actually have better oxygen saturation during REM than during NREM sleep. For example, in the ICSA or CSB-CSA syndromes, central apneas generally do not occur during REM sleep. Some patients with CompSA will also have a much better response to CPAP during REM sleep than during NREM sleep. TABLE 21–1 Effect of Sleep on Ventilatory Control A number of factors contribute to the occurrence of hypocapnic CSA (Table 21–2). Patients with hypocapnic CSA have a normal or low daytime PaCO2 and high ventilatory responses to hypercapnia. They develop central apnea because the PaCO2 falls below the apneic threshold (AT) to be discussed in the following section.14,15 From a ventilatory control model standpoint, they have high system loop gain and ventilatory instability.16,17 Central apneas are more common in stage N1 and N2 rather than stage N3 sleep because ventilation is less stable on transition from wakefulness to sleep. Sleep stage changes and arousal predispose to the occurrence of central apneas in patients with hypocapnic CSA.18 Hypocapnic central apneas are uncommon during REM sleep because ventilation is not totally under metabolic control and the ventilatory response to hypercapnia is lower than during NREM sleep, making ventilatory instability less likely. Short central apneas can occur during REM sleep during bursts of eye movements but these are likely not due to a lowering of PaCO2 below the AT. During wakefulness, hypocapnia does not cause cessation of breathing due to the presence of the wakefulness stimulus to breathing. During NREM sleep, ventilation depends completely on metabolic control. If the PaCO2 falls below a characteristic value (AT) for each individual, a central apnea occurs14,15 (Fig. 21–6). Ventilation does not resume until the PaCO2 climbs above (and actually slightly higher than) the AT. The AT can be experimentally determined by serial runs of hyperventilation with positive-pressure ventilation that progressively drop the PaCO2 to various levels below the eucapnic PaCO2 level during sleep. The positive-pressure ventilation is terminated suddenly, and if the PaCO2 has dropped below the AT, a central apnea occurs. For example, suppose the spontaneous sleeping PaCO2 is 42 mm Hg, then trials could progressively induce PaCO2 values of 41, 40, 39, 38, 37, and so on. The level at which a central apnea occurs when the positive-pressure device is turned off is the AT. As noted previously, the sleeping PaCO2 is normally about 2 to 8 mm Hg above waking value. The typical AT is usually at or 1 to 2 mm Hg lower than the waking PaCO2. In Figure 21–7, during normoxia, an individual end-tidal partial pressure of carbon dioxide (PETCO2) increases with sleep from 40 to 44 mm Hg. Hyperventilation trials with positive-pressure ventilation resulted in central apnea (apnea duration > 0 sec) in a range of PETCO2 values from about 38 to 42 mm Hg. Of note, it is the difference between the sleeping PaCO2 and the AT rather than the position of the AT that is the critical determinant of the propensity to develop central apnea. The smaller the PaCO2-AT difference the more likely CSA is to occur. Figure 21–8 shows awake and sleeping PaCO2 values for CHF patients with and without periodic breathing. Those with periodic breathing have a small difference between the sleeping PaCO2 and the AT. This is because the PaCO2 increased relatively little with transition from wake to sleep but the AT did not show a corresponding decrease.19 Studies have shown that the PaCO2-AT difference can vary with changes in ventilatory drive.20 Specifically, the PaCO2-AT difference increases with the induction of metabolic acidosis (acetazolamide) and decreases with metabolic alkalosis. This is somewhat counterintuitive because one might expect that further increases in ventilatory drive due to metabolic acidosis would increase the propensity for central apnea. In contrast to metabolic acidosis, hypocapnic hypoxia decreases the PaCO2-AT difference. Thus, hypoxemia can increase the likelihood of central apnea. A detailed description of the factors that affect the PaCO2-AT difference is found in references 15 and 20. Ventilatory control stability has also been analyzed using feedback control theory focusing on the loop gain (LG) of the respiratory system.2,16,17 The LG is determined by the plant gain (ability of increases in ventilation by the lungs/respiratory muscles to reduce the PaCO2) and the controller gain (change in ventilation induced by a change in PaCO2) (Fig. 21–9). High plant gain (Δ PaCO2/Δ ventilation) is associated with hypercapnia and low physiologic dead space. Owing to the hyperbolic relationship between ventilation and PaCO2, small changes in ventilation induce larger changes in PaCO2 if hypercapnia is present (Fig. 21–10A). Controller gain depends on the sensitivity of the ventilatory control centers to changes in PaCO2. Controller gain can be expressed as Δ Ventilation/Δ PaCO2. The LG = plant gain × controller gain. A system with a high LG (LG > 1) is unstable (see Fig. 21–10B). High controller gain or high plant gain can destabilize the system. In hypocapnic CSA, the major factors causing instability are the high controller gain and the effect of wake-to-sleep transitions. Both ICSA and CSB-CSA tend to be worse in the supine position.21,22 In ICSA patients, Issa and Sullivan21 found central apnea to be more frequent in the supine position. They hypothesized that upper airway reflexes may trigger central apnea because upper airway anesthesia abolished central apnea in two patients. In addition, high levels of continuous positive airway pressure (CPAP) abolished central apnea. CSB-CSA has also been noted to be more prominent in the supine position.22 It is not known whether this is due to upper airway factors or to changes in oxygenation or pulmonary congestion. The patterns of ventilation differ between patients with CSB-CSA and patients with ICSA. In both groups, the central apneas typically are 20 to 40 seconds in duration. In ICSA, there is typically a short ventilatory phase of 2 to 4 breaths between central apneas, and arousals tend to occur at apnea termination (Fig. 21–11). Patients with CSB have a longer ventilatory phase between consecutive apneas, and the ventilatory phase has a characteristic crescendo-decrescendo morphology. Hall and colleagues23 compared cycle length between patients with ICSA and CSB-CSA due to heart failure (Table 21–3). The apnea durations were similar but cycle length was longer in CSB-CSA due to a long ventilatory phase. The CSA-CSB patients also had a longer delay in the arterial oxygen saturation (SaO2) nadir. The lower the cardiac output, the longer the ventilatory phase and the delay in SaO2 nadir. This is believed due to a long circulation time. The patterns of ventilation in treatment-emergent CSA (nonhypercapnic, non-CSB CompSA) resembles that of ICSA. TABLE 21–3 Differences in Respiration between Patients with Idiopathic Central Sleep Apnea and Cheyne-Stokes Breathing From Hall MJ, Xie A, Rutherford R, Bradley TD: Cycle length of periodic breathing with and without heart failure. Am J Respir Crit Care Med 1996;154:376–381. These patients have central apneas of unknown etiology. CHF, neurologic disorders, or medications suppressing ventilation are not present. The prevalence varies but is thought to be 5% to 10% of patients being studied in sleep centers. Patients may present with symptoms similar to those of patients with OSA. These may include daytime sleepiness or insomnia, snoring, witnessed breathing pauses, and disturbed sleep. They tend to be thinner than patients with OSA and have less prominent snoring. In the ICSD-2, the terminology for this patient group is primary CSA (Box 21–4). As reflected in the term “idiopathic,” the etiology of primary CSA is unknown. However, the patients have ventilatory instability due to a high hypercapnic ventilatory response24 or sleep state instability.18 These patients tend to have decreased PaCO2 values during wakefulness. Often, central apnea can be triggered by only one or two large breaths (see Fig. 21–1). The periods of increased ventilation triggering central apneas often are associated with arousal. Arousal may trigger a transient increase in ventilation and a fall in PaCO2. This transient fall in PaCO2 is then associated with a central apnea as the patient returns to sleep. As discussed previously, in some patients with ICSA, central apnea occurs mainly in the supine position.21 Studies have documented a response to CPAP in some ICSA patients.21,25 In research studies, the addition of dead space or inhalation of PaCO2 with the goal of increasing and/or stabilizing the PaCO2 has been shown to reduce central apnea in ICSA patients.26 However, these interventions have not been tried for long-term treatment nor are they practical. Because idiopathic CSA is rare, most treatment information comes from small case series (no randomized, controlled studies) and no long-term studies of the effectiveness of any treatment have been published. There is no uniform consensus about the best treatment for patients with ICSA. This group is heterogeneous, and treatment must be individualized (Box 21–5). Various respiratory stimulants have been tried as treatments for idiopathic CSA, with variable amounts of success.27–29 The best evidence is for use of acetazolamide (Diamox), which is a carbonic anhydrase inhibitor. Acetazolamide induces a metabolic acidosis and reduces the pH even if the PaCO2 also decreases slightly. DeBacker and associates27 studied the effects of acetazolamide 250 mg 1 hour before sleep after 1 month of treatment in ICSA patients. The apnea-hypopnea index (AHI) was reduced by about 50% and symptoms improved, although sleep efficiency was not significantly better.27 Another treatment approach is to decrease arousals and promote stage N3 sleep where ventilation is more stable and the PaCO2 likely slightly higher. Hypnotics including triazolam and zolpidem have been tried.30–32 A recent trial of zolpidem 10 mg in ICSA was reported by Quadri and coworkers.31 The central AHI decreased from 30 to 13.5/hr and subjective sleepiness as measured by the Epworth Sleepiness Scale improved (from 13 to 8). However, in some patients, obstructive events increased with hypnotic treatment. Of note, hypnotics are relatively contraindicated in hypercapnic CSA. As noted previously, CPAP also appears to be effective treatment in some patients with ICSA.21,25 The mechanisms by which CPAP works are unknown. Four possibilities are that nasal CPAP minimizes overshoots in PaCO2 after arousal, slightly increases the sleeping PaCO2 in patients who are hypocapnic at baseline, prevents high upper airway resistance from inducing arousals, or prevents significant negative upper airway pressure (which may trigger reflex central apnea). Patients who snore or have central apnea mainly in the supine position might be assumed to be the best candidates for CPAP treatment. Of note, one case report found that CPAP helped but bilevel positive airway pressure (BPAP) worsened CSA.33 BPAP may destabilize the system by augmenting ventilation. No study evaluating the effect of adaptive servo-ventilation (ASV), a mode designed to stabilize ventilation, in ICSA has been published but a trial of that treatment might be reasonable. Supplemental oxygen has been shown to improve CSA in patients with CSB.34,35 However, an evaluation of supplemental oxygen in ICSA patients has not been published. Supplemental oxygen might work by decreasing ventilatory instability by blunting the hypercapnic ventilatory response. The hypercapnic ventilatory response is increased by hypoxia and decreased by hyperoxia. CSB occurs most commonly in patients with left ventricular systolic dysfunction36–38 but also can occur in patients with diastolic CHF or neurologic disorders.39,40 The neurologic disorders associated with CSB include a prior cerebrovascular accident (CVA) and neurodegenerative disorders. CSB may be present in up to 30% of patients in the first few days after stroke.39,41 Central apnea tends to resolve and is less commonly seen in studies of patients studied several months after stroke. OSA is the predominant form of sleep apnea present in patients after CVA. Of interest, a recent study suggested that a significant proportion of CSB-CSA in stroke patients was due to occult cardiovascular disease.42 CSA-CSB has also recently been reported in patients with heart failure with normal systolic function (diastolic heart failure).40 CSB-CSA due to systolic CHF is probably the most common cause of CSA seen today. Javaheri and colleagues36 found occult sleep-disordered breathing in 45% of a group with stable heart failure and an ejection fraction less than 45% and CSA was common in the affected individuals. MacDonald and associates37 studied a group of patients in stable CHF with maximal modern medical management and found 61% to have some form of sleep-disordered breathing (31% central apnea and 30% OSA). Oldenberg and coworkers38 studied 700 patients with CHF and found similar results. It should be noted that many patients in these studies had various amounts of both central and obstructive apneas. Defining what percentage of events qualified the patient for the central apnea group was somewhat arbitrary. In addition, patients can vary in the percentage of central events overnight43 or from night to night.44 In any case, the prevalence of CSB-CSA is very high in patients with significant systolic heart failure. Patients with CSA-CSB may complain of the typical symptoms of sleep apnea including disturbed sleep or daytime sleepiness. However, in most studies, the majority of patients do not complain of subjective excessive daytime sleepiness. Thus, a high index of suspicion is needed to suspect the presence of CSB. Nocturnal sleep complaints are often assumed to be secondary to CHF rather than to co-morbid sleep apnea. Despite the lack of subjective sleepiness complaints in CSB-CSA patients, studies have documented improvement in sleep quality and objective daytime sleepiness with successful treatment.45 The crescendo-decrescendo morphology of CSB-CSA has previously been discussed (see Figs. 21–2, 21–4, and 21–11). Another characteristic to appreciate is the long delay in the nadir in the SaO2 tracing after event termination due to a prolonged circulation time (low cardiac output) (Fig. 21–12). The distinctive pattern of CSB may not be recognized during a diagnostic study in a patient who has both OSA and underlying CSB.10 However, during a subsequent PAP titration, pure CSB-CSA may emerge when upper airway obstruction is eliminated (Fig. 21–13). The possibility of underlying CSB should be considered if mixed apneas during the diagnostic study have a long central component or if the nadir in the SaO2 following obstructive event termination is delayed. If central hypopneas rather than central apneas are present, this may also make CSB more difficult to recognize. As noted previously, central hypopneas rather than apneas can occur at the nadirs in ventilatory effort (see Fig. 21–2). The typical CSB cycle time is relatively long at 60 to 90 seconds and may be missed if a short time window is used to monitor sleep during a PAP titration. Figure 21–13 also illustrates the typical cessation of CSB-CSA when a patient transitions from NREM to REM sleep. The factors contributing to CSB include high ventilatory drive (high sympathetic tone, pulmonary congestion), long circulation time, and a small difference between the sleeping PaCO2 and the AT. Patients with CSB have low daytime PaCO2 compared with CHF patients without CSB46,47 due to increased ventilatory drive. High drive is thought to be due to increased sympathetic tone and pulmonary congestion stimulation of lung J receptors. In one study of patients with CHF, the higher the pulmonary capillary wedge pressure, the lower the awake PaCO2 (Fig. 21–14A).48 In another study, the higher the pulmonary capillary wedge pressure, the higher the AHI (see Fig. 21–14B).49 As noted previously, CHF patients with CSB also have a small difference between the sleeping PaCO2 and the AT.19
Central Sleep Apnea and Hypoventilation Syndromes
Classification of Csa Syndromes
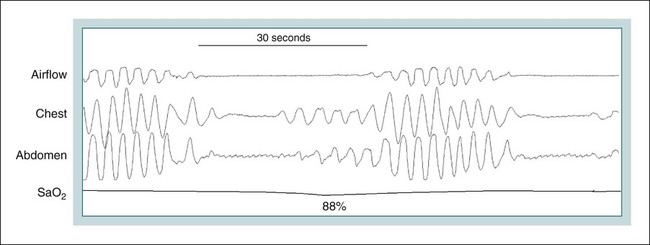
Pathophysiology of Csa
Effects of Normal Physiologic Changes
WAKE
NREM
REM
Wakefulness drive
Present
Absent
Absent
Central chemoreceptors (H+, PCO2)
Intact
Reduced
Very reduced
Peripheral chemoreceptors (PO2, PCO2 [H+])
Intact
Reduced
Very reduced
Apneic threshold
Not present
Present
N/A
Behavioral and other nonventilatory control center influences
Present
Absent
Periods of reduced tidal volume during phasic REM sleep often associated with bursts of eye movements
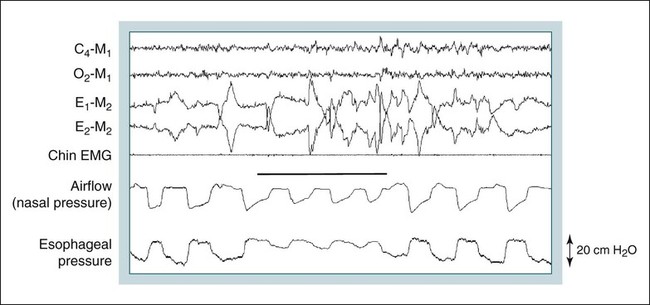
Ventilatory Control in Hypocapnic CSA
Apneic Threshold
Loop Gain
Upper Airway and Posture Effects
Hypocapnic Csa Syndromes
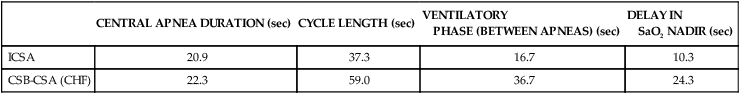
Patterns of Ventilation in ICSA and CSB
CENTRAL APNEA DURATION (sec)
CYCLE LENGTH (sec)
VENTILATORY
PHASE (BETWEEN APNEAS) (sec)
DELAY IN
SaO2 NADIR (sec)
ICSA
20.9
37.3
16.7
10.3
CSB-CSA (CHF)
22.3
59.0
36.7
24.3
Idiopathic CSA (Primary CSA)
Pathophysiology of Idiopathic CSA
Treatment of ICSA
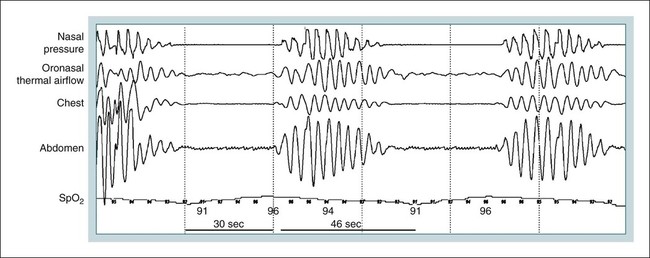
Csb with Csa
Polysomnography in CSB-CSA
Pathophysiology of CSB-CSA
Related posts:
 Sleep and Neurologic Disorders
Parasomnias
Sleep and Nonrespiratory Physiology—Impact on Selected Medical Disorders
Sleep Architecture Parameters, Normal Sleep, and Sleep Loss
The Technology of Sleep Monitoring: Differential Amplifiers, Digital Polysomnography, and Filters
Cardiac Monitoring during Polysomnography
Sleep and Neurologic Disorders
Parasomnias
Sleep and Nonrespiratory Physiology—Impact on Selected Medical Disorders
Sleep Architecture Parameters, Normal Sleep, and Sleep Loss
The Technology of Sleep Monitoring: Differential Amplifiers, Digital Polysomnography, and Filters
Cardiac Monitoring during Polysomnography
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Central Sleep Apnea and Hypoventilation Syndromes