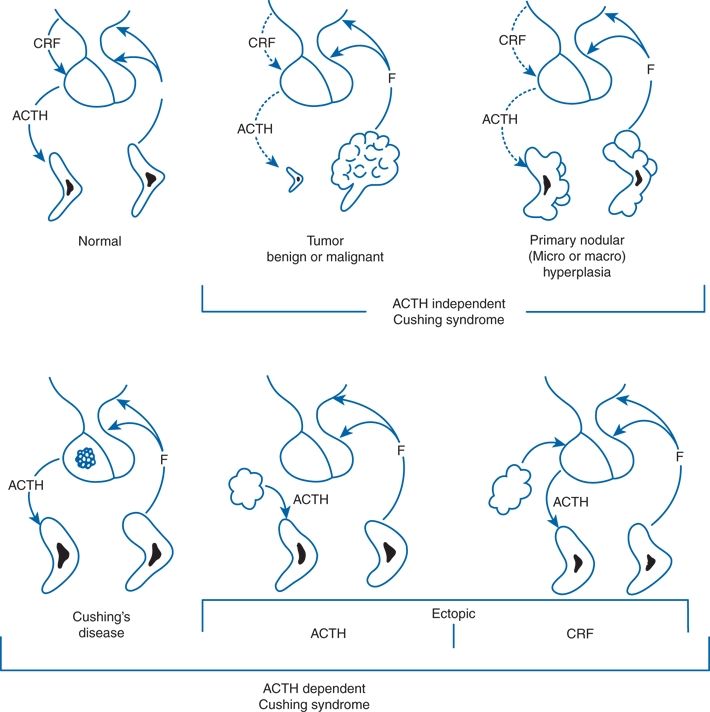
FIGURE 13-1 Causes of endogenous Cushing syndrome. The lesions on the top arise within the adrenal. Those in the bottom arise within the pituitary (Cushing disease) or from ectopic production of ACTH or corticotropin-releasing factor (CRF). F, cortisol. (Modified from Carpenter PC. Diagnostic evaluation of Cushing syndrome. Endocrinol Metab Clin NA 1988;17:445–472.)
ACTH-independent forms are mostly benign adrenal adenomas or malignant carcinomas, but various forms of hyperplasia may pose diagnostic difficulty. As noted in Chapter 12, the number of adrenal tumors found incidentally by abdominal CT or MRI is increasing. As many as 20% of these adrenal incidentalomas when initially recognized secrete cortisol in a partially unregulated manner, often in association with hypertension, diabetes, and generalized obesity (Rossi et al., 2000). Over 5 years, as many as 7% of those with initially normal cortisol regulation develop subclinical hyperfunction (Barzon et al., 2002). Adrenalectomy may be indicated for some with clinical features but “subclinical” hormonal tests (Mitchell et al., 2007).
A number of interesting variants have been reported, including
- Spontaneously remitting disease (Ishibashi et al., 1993)
- Cyclic or periodic disease (Manenschijn et al., 2012)
- Association with overt hypothalamic disorders (Dubois et al., 2007)
- Transition from pituitary-dependent to pituitary-independent disease (Hermus et al., 1988)
- ACTH-independent bilateral macronodular hyperplasia, which is often massive (Doppman et al., 2000), may be genetic (Beuschlein et al., 2014; Faucz et al., 2014), and associated with the expression of ectopic receptors for various hormones including the gastric inhibitory polypeptide (GIP), vasopressin, β-adrenergic agonists, LH/human CG or serotonin 5-HT4 (Bertherat et al., 2005; Lacroix et al., 2001). Such receptors are occasionally found in adrenal adenomas as well
- Pigmented micronodular dysplasia, in most cases as part of the autosomal dominant familial syndrome with cardiac and skin myxomas, the Carney complex (Bram et al., 2014)
- Association with pheochromocytoma (Lee et al., 2008), chemodectoma, carcinoid tumors (Corsello et al., 2014) and multiple endocrine neoplasia type 1 (Simonds et al., 2012).
- Increased sensitivity of peripheral glucocorticoid receptors without increased levels of cortisol (van Rossum & Lamberts, 2004)
Hypertension with Glucocorticoid Excess
Hypertension is present in about 75% of patients with Cushing syndrome. The severity of the hypertension may be related to the abolition of the normal nocturnal fall in BP seen after exogenous glucocorticoid administration and in patients with Cushing syndrome (Zelinka et al., 2004). The longer the duration of hypertension, the greater the likelihood that it will persist after relief of the syndrome (Suzuki et al., 2000).
Hypertension is relatively rare in patients who take exogenous glucocorticoids because of the use of steroid derivatives with less mineralocorticoid activity than cortisol. However, significant rises of BP can occur within 5 days of the administration of cortisol in fairly high doses (Whitworth et al., 2000).
Mechanisms for the Hypertension
Multiple mechanisms may be responsible for the hypertension so common in Cushing syndrome. The mechanisms may include the following:
- A sodium-retaining action of the high levels of cortisol. Although cortisol is 300 times less potent a mineralocorticoid than is aldosterone, 200 times more cortisol is normally secreted; this level is increased by two times or more in Cushing syndrome. With high levels of cortisol, the 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) capacity to convert cortisol to cortisone is overwhelmed, allowing cortisol to act on MCRs (Quinkler & Stewart, 2003; Ulick et al., 1992b).
- Glucocorticoids directly activate glucocorticoid receptors on vascular smooth muscle to raise blood pressure in knockout mice (Goodwin et al., 2008) and stimulate mineralocorticoid signaling in vascular smooth muscle cells in vitro, independent of aldosterone levels (Molnar et al., 2008).
- Increased production of mineralocorticoids. Though usually noted only in patients with adrenal tumors, increased levels of 19-nor-DOC (Ehlers et al., 1987), DOC, and less commonly, aldosterone (Cassar et al., 1980) have been found in patients with all forms of the syndrome.
- Stimulation of glucocorticoid receptors in the dorsal hindbrain (Scheuer et al., 2004).
- Reduced activity of various vasodepressor mechanisms (Saruta, 1996) in particular endothelial nitric oxide (Mangos et al., 2000).
- Increased levels of renin substrate and an increased responsiveness to various pressors (van der Pas et al., 2014).
- Other mechanisms may also be involved including an increase in erythropoietin (Whitworth et al., 2000) or endothelin (Kirilov et al., 2003).
Clinical Features
Many more patients with cushingoid features are seen than the relatively few who have the syndrome. The syndrome is more likely in patients with the clinical features shown in Table 13-2 (Prague et al., 2013) along with a hypercoaguable state (van der Pas et al., 2012). Significant hypokalemia is usually seen with the ectopic ACTH syndrome (Torpy et al., 2002). Significant hypokalemia is usually noted with the ectopic ACTH syndrome (Torpy et al., 2002).
TABLE 13-2 Clinical Features of Cushing Syndrome
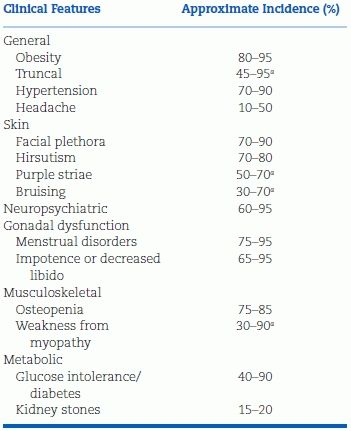
aMost discriminatory features.
Modified from Nieman LK et al. The diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 2008;93(5):1526–1540.
Cushing syndrome in children is usually manifested by weight gain and growth retardation, with systolic hypertension noted in 93% of 63 young patients (Magiakou et al., 1997). Fortunately, they usually become normotensive within a few months of surgical cure, but may have residual hypertension-related adverse effects (Lodish et al., 2009).
Pseudo-Cushing Syndrome
As many as 50% to 80% of patients with Cushing syndrome meet the criteria for major depression and may have persistent psychological and cognitive problems even after surgical remission (Resmini et al., 2012). On the other hand, patients with endogenous depression without Cushing syndrome may have poorly suppressible hypercortisolism related to increased ACTH pulse frequency (Mortola et al., 1987), but their basal cortisol levels are usually normal and they do not hyperrespond to corticotrophin-releasing hormone (CRH) (Yanovski et al., 1998).
Alcoholics often display numerous features suggestive of Cushing syndrome, including hypertension and elevated cortisol secretion (Badrick et al., 2008), which likely reflects increased secretion of corticotrophin-releasing factor (Groote Veldman & Meinders, 1996). On the other hand, 20% of patients with Cushing syndrome have hepatic steatosis by CT scans (Rockall et al., 2003).
Pregnant women often have features suggestive of Cushing syndrome; the rare appearance of Cushing syndrome during pregnancy may pose diagnostic dilemmas (Solomon & Seely, 2006).
Laboratory Diagnosis
Two somewhat contradictory scenarios exist in relation to the diagnosis of Cushing syndrome. First, the disease is being looked for in more patients with suggestive clinical features such as poorly controlled obese diabetics; in one study, 5.5% were found to have Cushing syndrome (Tabarin and Perez, 2011). This scenario requires screening tests with high specificity, i.e., few false positives—so that fewer suspects will have to be put through extensive confirmatory testing (Newell-Price, 2008).
The second scenario relates to the usually long duration between onset of symptoms and the time of diagnosis, averaging 6.0 years from one center (Psaras et al., 2011). This scenario requires confirmatory tests with high sensitivity, i.e., few false negatives—so that all patients can be correctly identified as early as possible. In view of the serious nature and the often irreversibility of the complications of the disease, the best balance is likely to be with a number of tests done over a short interval to achieve maximal predictive power (Findling & Raff, 2006). Nonetheless, controversy persists as to the appropriate cutoff values for different tests to provide the best balance (Newell-Price, 2008).
Screening Tests
For interpretation of cortisol levels, 1 μg/dL = 27 mmol/L.
The extent of the workup of patients suspected of having Cushing syndrome varies with the clinical situation. An overnight 1-mg dexamethasone suppression test (OST) will be adequate for most patients with only minimally suggestive features; patients with highly suggestive features should have repeated measurement of 24-hour urinary free cortisol (UFC) and midnight serum or salivary cortisol (Arnaldi et al., 2003; Findling & Raff, 2006; Nieman et al., 2008) (Table 13-3). In data from 4,126 patients, (Pecori et al., 2007) found the midnight serum cortisol, using a 1.8 μg/dL (50 mmol/L) cutoff, to provide only a 20% specificity whereas both the UFC and OST had 91% specificities. Nonetheless, the ease of obtaining the sample and the high sensitivity of the late-night salivary control lead Elias et al. (2014) to recommend it as the best screening study. The Endocrine Society recommends any one of the three screening tests: urine cortisol, late-night salivary cortisol, or 1-mg overnight dexamethasone suppression (Nieman et al., 2008) in keeping with their overall similar accuracy (Elamin et al., 2008).
TABLE 13-3 Tests to Determine the Anatomical Cause of Cushing Syndrome
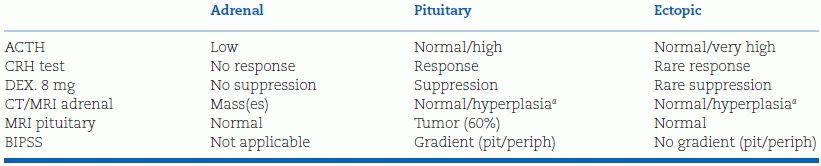
aNodules.
Dex, dexamethasone; DST, dexamethasone suppression test; BIPSS, bilateral inferior petrosal sinus sampling.
Data from Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: A consensus statement. J Clin Endocrinol Metab 2003;88(12):5593–5602.
Urinary Free Cortisol
The 24-hour UFC provides an integrated measure of the unbound circulating cortisol. High-performance liquid chromatography coupled with mass spectrometry provides better specificity than do immunoassays. The upper range of normal is 40 to 50 μg/24 hours (1,100 to l,380 mmol/L) and a value four times greater is usually diagnostic (Findling & Raff, 2006). The level may be lowered in patients with renal damage or raised with increased urinary volumes by reducing the fraction of filtered cortisol that is metabolized to cortisone or reabsorbed. Since the level is variable, three daily specimens are usually assayed.
Overnight Plasma Suppression
For screening, the single bedtime 1-mg dose dexamethasone suppression test (OST), measuring the plasma cortisol at 8 a.m. the next morning, has worked well but, to provide adequate sensitivity, the cutoff value should be 1.8 μg/dL, rather than the previously recommended 5 μg/dL (Findling et al., 2004). However, at the lower level, false-positive results are seen in about 10% of non-Cushing patients and false-negative results are seen in about 20% of patients with Cushing disease (Findling & Raff, 2006).
Late-Night Salivary Cortisol
An elevated late-night serum or salivary cortisol level is the earliest and most sensitive marker for Cushing syndrome (Elias et al., 2014). Rather than the inconvenience of obtaining blood samples, measurement of salivary cortisol levels in easily obtained samples has rapidly been accepted as a valid screening test including in children (Batista et al., 2007). Levels above 0.3 μg/dL (8.6 mmol/L) are abnormal (Findling & Raff, 2006).
Low-Dose Dexamethasone Suppression Test and Combined DST-CRH
DSTs may give anomalous results because hormone hypersecretion may be cyclic or variable. Pseudo-Cushing’s states, including depression, may be more accurately excluded by adding a CRH stimulation test after completion of the low-dose dexamethasone test (Yanovski et al., 1998). However, Gatta et al. (2007) found no additional diagnostic accuracy by the addition of CRH stimulation to the DST using a cutoff of plasma cortisol 15 minutes after CRH of 4 μg/dL (110 mmol/L). The plasma cortisol level value 15 minutes after CRH (1 μg/kg) is above 1.4 μg/dL (40 mmol/L) in patients with Cushing syndrome but remains suppressed in normals and patients with pseudo-Cushing’s.
Establishing the Cause of Cushing Syndrome
Once Cushing syndrome has been diagnosed, the anatomic cause needs to be accurately determined to guide therapy (see Table 13-3). In view of all the clinical vagaries and laboratory pitfalls that often confuse the differential diagnosis of the etiology of Cushing syndrome, referral to a medical facility with experience in dealing with such patients is almost always appropriate.
Corticotropin (ACTH) Assay
Measurement of plasma ACTH is the first step, using two-site immunometric assays that are sensitive, specific, and reliable, able to reliably detect values below 10 pg/mL (2 pmol/L). A suppressed ACTH concentration, below 5 pg/mL, indicates adrenal-independent Cushing syndrome usually from an adrenal tumor. However, other stimuli of adrenocortical receptors, such as insulinotropic peptide and vasopressin, may induce bilateral nodular adrenal hyperplasia with suppressed plasma ACTH levels. Normal or elevated plasma ACTH, above 20 pg/mL, indicate ACTH-dependent Cushing syndrome from either a pituitary or ectopic tumor. When values are between 10 and 20 pg/mL, a CRH stimulation test is indicated (Arnaldi et al., 2003).
Corticotrophin-Releasing Hormone Stimulation Test
Most pituitary tumors respond to IV CRH (1 μg/kg) with a release of plasma ACTH whereas adrenal tumors do not. Unfortunately, some ectopic ACTH-secreting tumors express the CRH receptor and also respond. Findling and Raff (2006) recommend a CRH test in patients with Cushing syndrome whose plasma ACTH levels are at the low end. The ACTH response is usually exaggerated if the pituitary tumor expresses the CRH receptor but blunted with adrenal tumors.
High-Dose Dexamethasone Suppression
Using the criterion of suppression of UFC to less than 10% of baseline for the diagnosis of pituitary-dependent Cushing disease, the high-dose (2 mg four times a day for 2 days) DST provides 70% to 80% sensitivity and close to 100% specificity (Boscaro et al., 2001). However, the results do not clearly separate ectopic ACTH from pituitary tumors, and this test is no longer recommended (Findling & Raff, 2006).
Pituitary MRI
In most patients, the measurement of plasma ACTH will be followed by a pituitary MRI with gadolinium enhancement (Lonser et al., 2013). Thereby, a discrete pituitary adenoma will be seen in about 60% of patients; if the tumor is greater than 6 mm in size, no further studies are required and the patients may be referred to a pituitary neurosurgeon (Arnaldi et al., 2003). It should be remembered that almost 15% of the general population harbor incidental pituitary tumors, although most are below 5 mm in diameter (Karavitaki, 2007). Since some patients with an ectopic ACTH-secreting tumor have abnormal pituitary MRI findings, bilateral interior petrosal sinus sampling is indicated in those with clinical features suggesting an ectopic tumor, such as rapid onset of symptoms or hypokalemia (Findling & Raff, 2006).
Inferior Petrosal Sinus Sampling
Bilateral simultaneous sampling of the inferior petrosal sinuses (IPSS) is a powerful means of confirming whether or not the source of corticotropin is the pituitary, especially if imaging is negative. Ratio of central to peripheral ACTH of greater than 3 after CRH stimulation provides a sensitivity of 95% to 97% and specificity of 100% in diagnosing pituitary-dependent Cushing disease (Arnaldi et al., 2003). Less discrimination was found in a series of 185 IPSS procedures with a 99% positive predictive power but only a 20% negative predictive power (Swearingen et al., 2004). In view of the technical difficulty with IPSS, sampling of the internal jugular vein may be performed and only patients with a negative result referred for IPSS (Ilias et al., 2004).
If clinical and lab data point to an ectopic ACTH-secreting tumor, CT and/or MRI of the neck, thorax, and abdomen and, for occult tumors, scintigraphy with the somatostatin analog, 111In-pentetreotide, are currently used to locate the tumor (De Herder & Lamberts, 1999). Positron emission tomography with other labeled precursors has identified ACTH-secreting carcinoid tumors (Dubois et al., 2007).
Treatment
Treatment of the Hypertension
Until definitive therapy is provided, the hypertension that accompanies Cushing syndrome can temporarily be treated with the usual antihypertensive agents described in Chapter 7. Since excess fluid volume is likely involved, a diuretic, perhaps in combination with an aldosterone antagonist, spironolactone or eplerenone, is an appropriate initial choice. After definitive therapy, hypertension usually improves but coronary disease risk factors often persist, likely because of residual abdominal obesity and insulin resistance (Barahona et al., 2013).
Treatment of the Syndrome in General
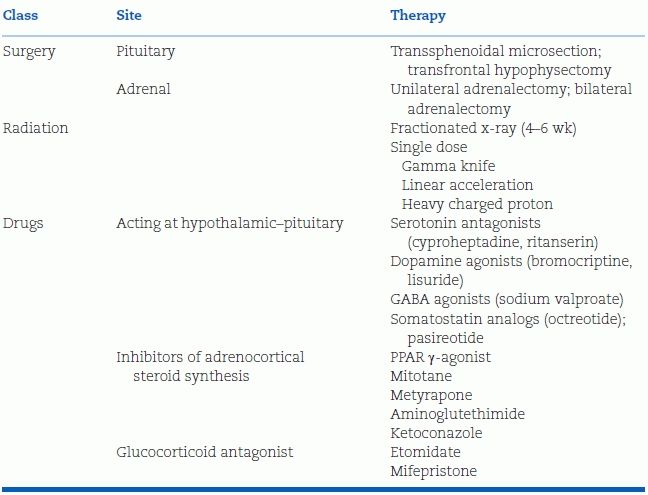
In view of the long-term morbidity associated with Cushing syndrome, the condition must be treated as rapidly as possible after the diagnosis has been established (Pulse et al., 2013). The choice of definitive therapy depends on the cause of the syndrome (Table 13-4).
- In the majority of patients who have ACTH-dependent Cushing disease with a pituitary tumor, transsphenoidal microsurgical removal is the treatment of choice (Hassan-Smith et al., 2012; Wagenmakers et al., 2013). In some circumstances, bilateral adrenalectomy or stereotactic radiotherapy (Petit et al., 2008) may be used if pituitary surgery is unsuccessful or when no pituitary tumor is found (Pouratian et al., 2007).
- Benign adrenal tumors should be surgically removed, increasingly by laparoscopy (Conzo et al., 2014).
- For adrenal cancers and ectopic ACTH tumors that cannot be resected, removal of the adrenal may be helpful, but chemotherapy is usually needed (Pozza et al., 2012).
- The drugs listed in Table 13-4 are mainly used to quickly overcome severe complications, either in preparation for surgery or whenever definitive treatment must be delayed (Trainer, 2014).
TABLE 13-4 Therapies for Cushing Syndrome
Follow-up
With definitive therapy, remission rates of 70% to 80%—defined as normal plasma and urinary cortisol levels and resolution of clinical stigmata—have been noted (Arnaldi et al., 2003; Hassan-Smith et al., 2012). However, as many as 25% of pituitary-dependent Cushing’s patients have recurrences at 5 years after initially successful transsphenoidal surgery, so close and long-term follow-up is necessary (Alexandraki et al., 2013).
SYNDROMES WITH INCREASED ACCESS OF CORTISOL TO MINERALOCORTICOID RECEPTORS
Less common than Cushing syndrome caused by cortisol excess are a variety of fascinating syndromes wherein normal or increased levels of cortisol exert a mineralocorticoid effect by binding to the renal MCRs. As depicted in Figure 13-2, the normal renal MCR is as receptive to glucocorticoids as it is to mineralocorticoids. The 11β-hydroxysteroid dehydrogenase type 2 isoform (11β-HSD2) enzyme in the renal tubules upstream to these receptors normally converts the large amounts of fully active cortisol to the inactive cortisone, thereby leaving the MCRs open to the effects of aldosterone (Quinkler & Stewart, 2003).
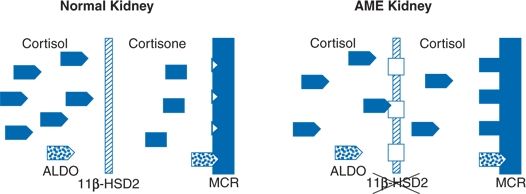
FIGURE 13-2 Enzyme-mediated receptor protection. Normally, 11β-dehydrogenase (11β-HSD2) converts cortisol to inactive cortisone in the more proximal nephron, protecting mineralocorticoid receptors (MCRs) from cortisol and allowing selective access for aldosterone. When 11β-HSD2 is defective, e.g., in congenital deficiency (AME kidney) or after licorice administration, cortisol gains inappropriate access to mineralocorticoid receptors, resulting in sodium retention and potassium wasting. (Modified from Cerame BI, New MI. Hormonal hypertension in children: 11β-Hydroxylase deficiency and apparent mineralocorticoid excess. J Ped Endocrinol Metab 2000;13:1537–1547.)
However, there are both congenital and acquired deficiencies of the 11β-HSD2 enzyme, so that the normal levels of cortisol remain fully active, flooding the MCR and inducing the full syndrome of mineralocorticoid excess: Sodium retention, potassium wastage, and hypertension with virtually complete suppression of renin and aldosterone secretion (Stewart, 2003).
11β-HSD2 Deficiency: Apparent Mineralocorticoid Excess
Apparent mineralocorticoid excess (AME) is an autosomal recessive disorder that has now been identified in about 100 patients. The syndrome clinically is characterized by familial consanguinity, low birth weight, failure to thrive, onset of severe hypertension in early childhood with extensive target organ damage, hypercalciuria, nephrocalcinosis, and renal failure (Chemaitilly et al., 2003). As noted, sodium retention, hypokalemia, low aldosterone, and low renin levels are present.
Genetics
Soon after the first case was described (Werder et al., 1974), Ulick et al. (1979) recognized that these children did not metabolize cortisol normally. Some years later, Stewart et al. (1988), in studies on a 20-year-old with the syndrome, recognized a defect in the renal cortisol–cortisone shuttle and demonstrated the deficiency of the 11β-HSD2 enzyme. A number of mutations in the 11β-HSD gene have now been identified in patients with AME (Carvajal et al., 2003; Friso et al., 2008).
Some of these mutations result in only partial inhibition of the 11β-HSD2 enzyme as evidenced by a higher ratio of urinary cortisone to cortisol metabolites and a milder clinical course with larger birth weight, later age of presentation (Nunez et al., 1999), and in at least one patient, only mild low-renin hypertension (Wilson et al., 1998). Not surprisingly, mutations resulting in less inhibition of the enzyme have been sought in patients with “essential” hypertension. Some have found them but most have not (Quinkler & Stewart, 2003). A role of impaired 11β-HSD2 activity has also been proposed for sodium sensitivity (Ferrari et al., 2001), intrauterine growth retardation (McTernan et al., 2001), and preeclampsia (Schoof et al., 2001).
An intriguing prospect has been proposed that decreased 11β-HSD2 activity may occur with aging and thereby may be involved in hypertension in the elderly (Campino et al., 2013).
Variant
A few patients with the features of AME have a defect not in the cortisol to cortisone shuttle but in the ring A reduction of cortisol to inactive metabolites because of a deficiency of the 5β-reductase enzyme (Ulick et al., 1992a). The resultant high levels of cortisol keep the MCRs flooded in the same manner as when 11β-HSD2 is deficient.
Therapy
Therapy is usually based on competitive blockade of the MCR with spironolactone (Dave-Sharma et al., 1998) or eplerenone (Funder, 2000). Suppression of endogenous cortisol with dexamethasone has also been used (Quinkler & Stewart, 2003). Cure has been reported on one patient after transplantation of a kidney with normal 11β-HDS2 activity (Palermo et al., 1998).
11β-HSD2 Inhibition: Glycyrrhetinic Acid (Licorice)
Since the early 1950s, glycyrrhizin acid, the active ingredient in licorice extract, has been known to cause hypertension, sodium retention, and potassium wastage. Stewart et al. (1987) and Edwards et al. (1988) recognized the similarities between the syndrome induced by licorice and the syndrome of AME and documented that licorice inhibited the same renal 11β-HSD2 enzyme that was deficient in AME. These effects are accompanied by a fall in cortisone and a rise in cortisol excretion, reflecting the inhibition of renal 11β-HSD2 activity.
Relatively small amounts of confectionary licorice, as little as 50 g daily for 2 weeks, produce a rise in BP in normal people (Sigurjonsdottir et al., 2001). The syndrome also has been induced by the licorice extracts in chewing tobacco and candy and herbal remedies (Sontia et al., 2008). Not surprisingly, aldosterone receptor blockers (spironolactone and eplerenone) have been shown to relieve all of the effects of licorice-induced hypertension (Quaschning et al., 2001). Even better is to recognize and stop the habit.
Massive Cortisol Excess
The capacity of the 11β-HSD–directed cortisol–cortisone shuttle and of 5β-reductase inactivation may be overcome by massive amounts of cortisol. Ulick et al. (1992b) have shown this to be the mechanism responsible for the significant features of mineralocorticoid excess—profound hypokalemia and hypertension—that are seen in patients with ectopic ACTH tumors wherein cortisol levels are much higher than in other causes of Cushing syndrome (Torpy et al., 2002).
Glucocorticoid Resistance
Both sporadic and familial forms of glucocorticoid receptor resistance, ascribed to various mutations in the receptor gene (Nicolaides et al., 2014), have increased levels of circulating cortisol but without typical Cushing stigmata (Kino et al., 2002). Many of these patients have hypertension that may mimic mineralocorticoid excess. Moreover, among 60 hypertensive patients under age 36, 45 had increased levels of urinary glucocorticoid metabolites, suggesting partial resistance of glucocorticoid receptors with subsequent increased mineralocorticoid effects (Shamim et al., 2001).
DEOXYCORTICOSTERONE EXCESS: CONGENITAL ADRENAL HYPERPLASIA
Excessive amounts of the mineralocorticoid DOC may cause hypertension (Ferrari & Bonny, 2003), arising either from hyperplastic adrenals with enzymatic deficiencies or from rare DOC-secreting tumors (Gröndal et al., 1990).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


