Chapter 62 Inborn Errors of Metabolism of the Nervous System
General Considerations
Diagnostic Approach
When an IEM is possible, the clinical features help focus the approach to diagnosis. In the setting of acute illness, consider an IEM in parallel with more common disorders, even with a noninformative family history. Examine the blood and urine of patients with acute neurological deterioration for signs of acidosis, ketosis, hypoglycemia, and hyperammonemia (Tables 62.1 and 62.2). Also consider screening tests for abnormalities of amino or organic acids. Abnormal metabolites may not be present during stable periods or in samples obtained after the acute illness is over.
Table 62.1 Commonly Requested Tests for the Evaluation of a Patient Thought to Have an Inborn Error of Metabolism
| Tests | Clinical Utility |
|---|---|
| Ammonia | Urea cycle defects, organic acidemia |
| Carnitine, plasma or serum total and free (unesterified) urine levels | Deficiency may develop in carnitine transport defects, disorders of fatty acid oxidation and branched-chain amino acid metabolism, and valproic acid treatment |
| Acylcarnitine profile | Normal plasma acyl/free carnitine ratio: <0.25 |
| Ceruloplasmin | Decreased in Wilson and Menkes disease, aceruloplasminemia |
| Cholesterol | Low plasma levels in Smith-Lemli-Opitz syndrome, cerebrotendinous xanthomatosis; abnormal profile in the dyslipidemias |
| Free fatty acids (FFA); ketone bodies (KB): 3-hydroxybutyrate, acetoacetate | Disorders of fatty acid oxidation and ketolysis; supervised fasting and assessment of FFA/KB ratio and glucose and ketone levels enable distinction of hypo- and hyperketotic disorder |
| Lactate* | Defects of glycogen metabolism, gluconeogenesis and fatty acid oxidation (often seen with hypoglycemia); defects involving the electron transport chain, Krebs cycle, and pyruvate dehydrogenase (absence of hypoglycemic episodes) Lactate/pyruvate ratio (NI: <20 : 1) provides insight into oxidation-reduction status (increased in OXPHOS defects; normal in PDHC deficiency) Normal blood lactate <1.8 mmol/L Normal CSF lactate <2.2 mmol/L |
| Very long-chain fatty acid (VLCFA) | Disorders of peroxisomal metabolism |
| Phytanic acid | Elevated in Refsum disease and rhizomelic chondrodysplasia punctata |
| Uric acid | Elevated in Lesch-Nyhan syndrome† and other defects of purine metabolism and glycogen storage disorders; decreased in molybdenum cofactor deficiency and defects of pyrimidine metabolism CSF/plasma ratio |
| Glucose | <0.35 Glucose transport defect |
| Glycine | >0.6 Nonketotic hyperglycinemia; low in plasma and CSF glycine levels in 3-phosphoglycerate dehydrogenase deficiency; plasma glycine markedly elevated in patients with organic acidemias (MMA, PA, IVA) |
| Serine | <0.2 Serine deficiency syndromes (3-phosphoglycerate dehydrogenase and phosphoserine phosphatase deficiency) |
CSF, Cerebrospinal fluid; IVA, isovaleric acidemia; MMA, methylmalonic acidemia; OXPHOS, oxidative phosphorylation; PA, propionic acidemia; PDHC, pyruvate dehydrogenase complex.
* Presence or absence of hypoglycemia can be a useful aid to differential diagnosis of disorders that lead to lactic acidemia.
† Hyperuricemia may lead to formation of nephrolithiasis, obstructive nephropathy, and gout.
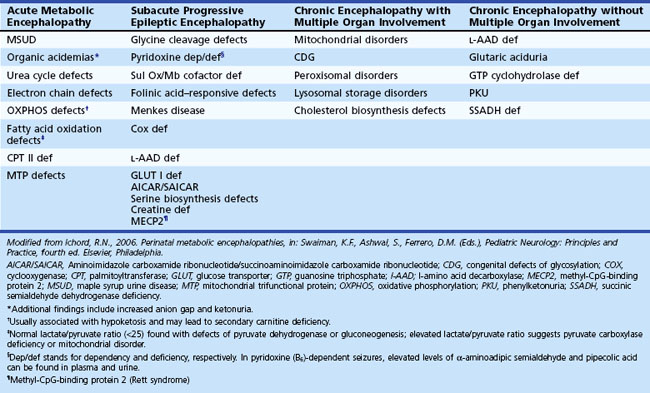
Certain situations require an analysis of cerebrospinal fluid (CSF) (Jones et al., 2006). For example, determination of CSF levels of biogenic monoamines and γ-aminobutyric acid (GABA) may be diagnostic in severe neonatal/infantile epileptic encephalopathy due to neurotransmitter defects (Table 62.3). Such defects are suggested in infants and children with (fluctuating) extrapyramidal disorders, in particular parkinsonism, dystonia or more general “athetoid cerebral palsy,” and vegetative disturbances. Other diagnoses for which CSF testing may be informative include nonketotic hyperglycinemia, GABA transaminase deficiency, and homocarnosinosis. Proper collection and handling of the CSF is important to avoid spurious results (Hyland, 2003).
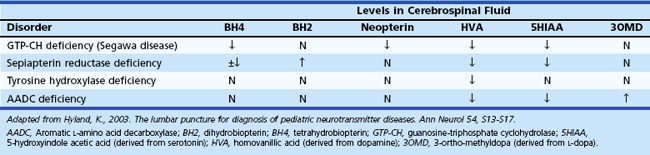
Neurological deterioration is a characteristic feature of acute intoxication disorders (e.g., certain aminoacidopathies [maple syrup urine disease, MSUD], organic acidemias [methylmalonic acidemia, MMA; propionic acidemia, PA; isovaleric acidemia, IVA; multiple carboxylase deficiency, MCD], and the urea cycle defects). Abnormal urine odor is present in diseases associated with the excretion of volatile metabolites (maple syrup in MSUD; sweaty feet in IVA and glutaric acidemia type II). Isolated seizures are often the initial features of vitamin-responsive disorders (e.g., defects of pyridoxine and folinic acid metabolism, biotin-responsive MCD), and are a prominent feature in nonketotic hyperglycinemia, sulfite oxidase deficiency, and congenital malabsorption of magnesium. Congenital lactic acidosis and central hypotonia are features of deficiencies of pyruvate carboxylase (PC) and pyruvate dehydrogenase (PDH) and disorders of the Krebs cycle and mitochondrial respiratory chain (Pithukpakorn, 2005). Recurrent hypoglycemia typically occurs in the glycogen storage disorders (GSD) affecting the liver and in defects of FAO (consequent to overutilization); these diseases are also often associated with signs of cardiac involvement (cardiomyopathy, arrhythmias). In the mitochondrial FAO defects, urinary ketone concentrations are low, and the ratio of 3-hydroxybutyrate/acetoacetate is usually less than 2.0. Analysis of plasma acylcarnitine and urinary acylglycine profile are particularly helpful in these cases. Renal tubular acidosis associated with HCO3− loss in urine can be encountered in PC deficiency, MMA, carbonic anhydrase deficiency, carnitine palmitoyltransferase I (CPT-I) deficiency, and cystinosis.
Ophthalmological examination often provides a clue to the diagnosis of IEM. Vertical supranuclear ophthalmoplegia occurs in Niemann-Pick disease type C, and saccadic initiation failure and defective optokinetic nystagmus occurs in Gaucher disease type III. Kayser-Fleisher rings (orange or greenish deposits around the limbus of the cornea due to copper deposition within the Descemet membrane) are a feature of Wilson disease. Table 62.4 shows additional ophthalmological findings characteristic of IEM.
Table 62.4 Ophthalmologic Findings Associated with Inborn Errors of Metabolism
| Ophthalmologic Abnormality | Associated Inborn Errors of Metabolism |
|---|---|
| Cataracts | Cerebrotendinous xanthomatosis |
| Cholesterol synthesis defects | |
| Galactokinase deficiency | |
| Galactosemia | |
| Lowe syndrome | |
| Menkes syndrome | |
| Mucopolysaccharidoses | |
| Peroxisomal disorders | |
| Serine deficiency disorders | |
| Tyrosinemia type II | |
| Cherry red spot | Galactosialidosis |
| GM1-gangliosidosis | |
| Niemann-Pick disease A | |
| Sandhoff disease | |
| Sialidosis type I | |
| Tay-Sachs disease | |
| Optic atrophy | Adrenoleukodystrophy |
| Canavan disease | |
| Hyperornithinemia with gyrate atrophy | |
| Lafora disease | |
| Lens dislocation | Homocystinuria |
| Molybdenum cofactor deficiency | |
| Sulfite oxidase deficiency | |
| Retinopathy | Carbohydrate-deficient glycoprotein syndrome |
| Neuronal ceroid lipofuscinosis | |
| Mitochondrial defects | |
| Peroxisomal disorders |
Tandem mass spectrometry (TMS) analysis of blood spots on filter paper is an effective means of screening for some defects of amino and organic acid metabolism and FAO defects (Pasquali et al., 2006) (Table 62.5). Methods for screening of lysosomal storage disorders by TMS have also been recently introduced (Matern, 2008). Metabolic profiling of infant urine through comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry for the diagnosis of organic acidurias and biomarker discovery is also an option (Kouremenos et al., 2009). The main advantages of TMS over previous methods of newborn screening are improved accuracy, sensitivity and specificity, and suitability for cost-effective multi-IEM screening. Furthermore, its use avoids the need for potentially harmful procedures (i.e., fasting, substrate loading).
Table 62.5 Metabolic Disorders Detected Through Tandem Mass Spectrometry
| Disorder | Enzyme Deficiency | Primary Metabolic Indicator |
|---|---|---|
| AMINO ACIDOPATHY | ||
| Phenylketonuria/hyperphenylalaninemia | Phenylalanine hydroxylase (and variants) | Phe |
| Maple syrup urine disease | Branched chain oxo- (or keto-) acid dehydrogenase | Leu/Ile, Val |
| Homocystinemia | Cystathionine β-synthase | Met |
| Hypermethioninemia | Methionine-S-adenosyltransferase | Met |
| Citrullinemia | Argininosuccinate synthetase | Cit |
| Argininosuccinic aciduria | Argininosuccinate lyase | Cit |
| Tyrosinemia type I | Fumarylacetoacetate hydrolase | Tyr |
| ORGANIC ACIDEMIA | ||
| Glutaric acidemia type 1 | Glutaryl-CoA dehydrogenase | C5DC |
| Propionic acidemia | Propionyl-CoA carboxylase | C3 |
| Methylmalonic acidemia | Methylmalonyl-CoA mutase | C3 |
| Isovaleric acidemia | Isovaleryl-CoA dehydrogenase | C5 |
| 3-Hydroxy-3-methylglutaryl CoA lyase deficiency | C5OH | |
| 3-Methylcrotonyl carboxylase deficiency | C5OH | |
| FATTY ACID OXIDATION DEFECTS | ||
| Medium-chain acyl-CoA dehydrogenase deficiency | C8, C10, C10:1, C6* | |
| Very long-chain acyl-CoA dehydrogenase deficiency | C14:1, C14, C16 | |
| Short-chain acyl-CoA dehydrogenase deficiency | C4 | |
| Multiple acyl-CoA dehydrogenase deficiency | C4, C5, C8, C12, C14, C16, C5DC | |
| Carnitine palmitoyltransferase deficiency | C16, C18:1, C18 | |
| Carnitine/acylcarnitine translocase deficiency | C160H, C18:1OH, C18OH | |
| Very long-chain hydroxyacyl-CoA dehydrogenase deficiency | C16OH, C18:1OH, C18OH | |
| Trifunctional protein deficiency | C16OH, C18:1OH, C18OH |
* The abbreviations for fatty acid oxidation products and organic acid intermediates (e.g., C5, C160H, C18:1, etc.) refer to carnitine esters of aliphatic monocarboxylic acids, with chain length indicated by the number adjacent to C, and the number of double bonds indicated by the number after the colon.
From Centers for Disease Control and Prevention, 2001. Using tandem mass spectrometry for metabolic disease screening among newborns. Recommendations and reports. MMWR Morb Mortal Wkly Rep 50, April 13, 2001.
Assessment of carnitine profile (total and esterified carnitine levels and urinary carnitine excretion patterns) may also prove useful when suspecting a primary or secondary carnitine deficiency. Carnitine plays an essential role in the transfer of long-chain fatty acids across the inner mitochondrial membrane, in the detoxification of acyl moieties, and in the maintenance of free coenzyme A levels. Dietary sources provide carnitine. Primary carnitine deficiency (due to defective transport protein encoded by the OCTN2 gene) leads to increased urinary loss and cardiac and skeletal muscle disease. Secondary carnitine deficiency occurs in several IEM and may be partially responsive to oral carnitine supplementation. Table 62.6 shows the differential diagnosis of disorders involving carnitine metabolism.
Table 62.6 Useful Measures to Aid in the Differential Diagnosis of Inborn Errors of Metabolism Involving Carnitine
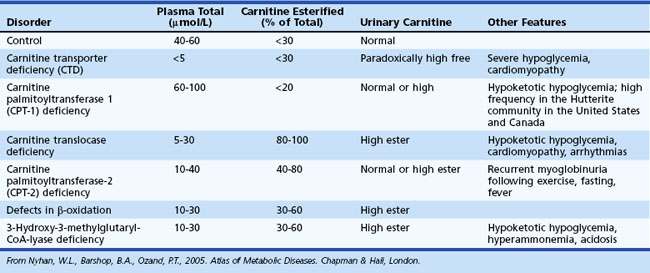
Histological examination of appropriate tissue samples can provide clues to the nature of the storage materials found in certain IEM (e.g., lysosomal disorders) (Warren and Alroy, 2000). When performing a skin biopsy, it is advisable to obtain samples for microscopic examination and tissue culture. These are useful as source material for subsequent biochemical or molecular (genetic) testing. Disorders of amino and organic acid metabolism are not associated with deposition of storage material, and histological findings are nonspecific. For this group of disorders, use skin fibroblasts for confirmatory diagnosis by enzymatic assays. For some disorders, biochemical testing is not accurate for carrier detection, because residual enzyme activity in a significant proportion of carriers overlaps with values obtained from the general population. In certain IEM, molecular assays may be available for diagnostic purposes and carrier testing. Microscopic examination of scalp hair can provide important diagnostic information in some cases, such as in Menkes syndrome (pili torti) (Smith et al., 2005).
The GeneTests website (www.genetests.org) lists diagnostic laboratories performing specialized genetic tests. Careful attention to sample requirements and shipping and handling considerations is imperative. Clinical information is required with the specimen to receive expert advice in patient evaluation. In cases with an established diagnosis, detailed information may be obtained from several websites, including Online Mendelian Inheritance in Man (OMIM [www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM]), GeneClinics (www.geneclinics.org), and the National Organization for Rare Disorders (NORD [www.rarediseases.org]). Several support and patient advocacy groups provide information about community resources and ongoing clinical trials.
Mutation Analysis in the Diagnosis of Inborn Errors of Metabolism
Examples of disorders for which DNA testing has proven useful include medium-chain acyldehydrogenase [MCAD] deficiency, myophosphorylase deficiency (McArdle disease), and Gaucher disease (Gregersen et al., 2004; Madonna et al., 2002; Maire, 2001). Among patients with MCAD deficiency of northwestern European descent, 80% are homozygous for a single missense mutation (A985G), and 17% carry this mutation in combination with another less common defect. This finding has improved the reliability of MCAD carrier identification and diagnosis, particularly of siblings who may be affected but asymptomatic at the time of family screening. Certain metabolic disorders (e.g., Tay-Sachs disease, Gaucher disease, Niemann-Pick disease type A, and Canavan disease) have an increased prevalence among individuals of Ashkenazi Jewish ancestry (i.e., of Central and Eastern European descent). A limited number of “common” mutations in this population cause the disease. This has enabled targeted screening for appropriate counseling prior to marriage. In hereditary fructose intolerance and dihydropyrimidinase deficiency, screening for the disease mutation in blood obviates the need for liver biopsy. However, in mitochondrial disorders that cause myopathy, biopsy of a weak muscle is still required, because expression of the mutant gene may not occur in other tissues. Fresh muscle tissue is also the preferred sample for studies of respiratory chain function.
Special Considerations
Deficiency of succinate dehydrogenase or fumarase activity can lead to lactic acidosis. Interestingly, heterozygous mutations involving the succinate dehydrogenase (SDH) complex have been associated with hereditary paraganglioma and pheochromocytoma. Heterozygous mutations involving fumarase have been associated with hereditary leiomyomatosis and renal cell cancer, a genodermatosis, and familial renal cancer syndrome (Pollard, 2003). Thus, parents and at-risk family members of patients with SDH or fumarase deficiency require screening for these tumors.
Recent studies suggest an increased incidence of parkinsonism in families segregating for Gaucher disease (Neumann et al., 2009).
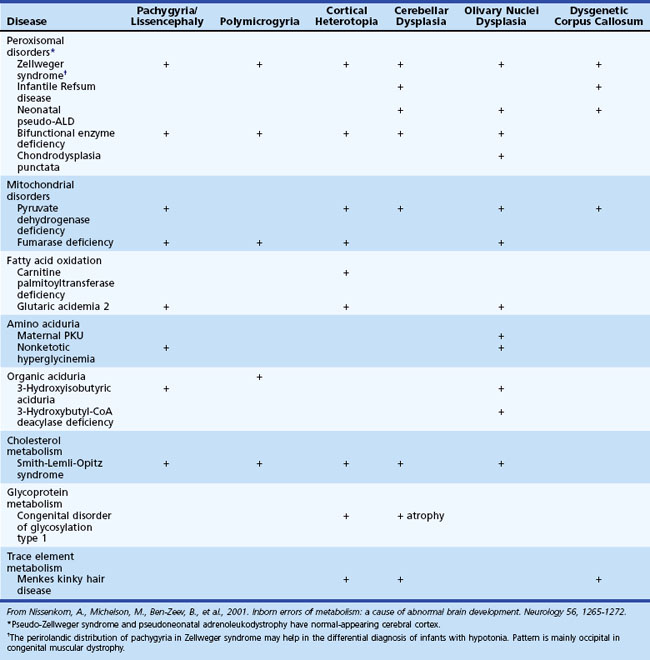
Inborn Errors of Metabolism Associated with Abnormal Brain Development and Encephaloclastic Lesions
Several metabolic disorders can cause a disruption of the normal sequence of brain development and lead to encephalocele, dysgenic corpus callosum, and neuronal migration defects (Hennekam, 2005; Jeng et al., 2001; Nissenkorn et al., 2001) (Tables 62.7 and 62.8). For instance, cystic necrosis of white matter, with or without basal ganglia involvement, occurs in PDH, PC, and molybdenum cofactor deficiencies. Nonsyndromic congenital microcephaly has been associated with maternal PKU, phosphoglycerate dehydrogenase deficiency, and 2-ketoglutaric aciduria (the cause of Amish lethal microcephaly). The proposed mechanisms to explain abnormal brain development and encephaloclastic lesions (such as porencephalic cysts) in IEM include the production of a toxic or energy-deficient intrauterine milieu, modification of the content and function of membranes, and disturbance of the normal expression of intrauterine genes responsible for neurulation and neuronal migration (Prasad et al., 2009).
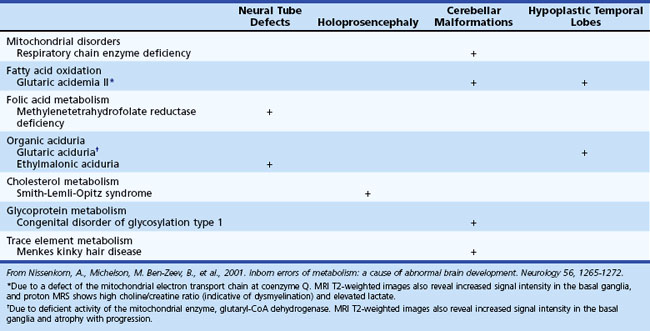
Neuroimaging in the Diagnosis and Management of Neurometabolic Brain Diseases
Magnetic resonance imaging (MRI) has been widely used to obtain brain anatomical data in patients with static and progressive encephalopathy. Advanced imaging techniques such as MR spectroscopy (MRS) and diffusion-weighted imaging (DWI) provide additional information relating to brain biochemistry and cell viability and point to the diagnosis of a neurometabolic disease (Cheon et al., 2002; Faerber and Poussaint, 2002; Kaye, 2001). MRS and DWI are quantitative techniques that may prove useful in assessing severity of the underlying pathological changes and potential response to treatment.
White-matter signal abnormalities consistent with leukodystrophy can be found in several metabolic disorders such as Krabbe disease, metachromatic leukodystrophy (MLD), X-linked adrenoleukodystrophy, and Canavan disease. In addition to signal hyperintensities noted on T2-weighted imaging, special features may provide additional clues to the diagnosis. Enlarged perivascular spaces or small cysts are characteristically noted in the mucopolysaccharidoses and Lowe syndrome. Gray-matter lesions may be found in Zellweger syndrome and other peroxisomal disorders. In glutaric aciduria type 1, imaging may show selective frontotemporal atrophy, especially involving subcortical white matter, with prominent extraaxial CSF collection, widening of the sylvian fissure with poor opercularization (“bat wing” appearance), and in some cases, subdural hemorrhage (Oguz et al., 2005). The presence of T2 hyperintense lesions in the subcortical cerebral white matter, basal ganglia, and dentate nuclei, together with cerebellar atrophy, is considered pathognomonic of l-2-hydroxyglutaric aciduria (Steenweg et al., 2009).
MRS has enabled definition of the creatine deficiency syndromes, a newly discovered group of disorders causing mental retardation and other neurological problems (i.e., extrapyramidal movement abnormalities, hypotonia). The common feature is severe depletion of brain creatine/phosphocreatine. With brain MRS, elevated brain levels of N-acetylaspartate (NAA) are seen in patients with Canavan disease, whereas patients with mitochondrial defects, defects of gluconeogenesis, and biotin-responsive multiple carboxylase deficiency may show elevated brain lactate levels. In MLD, MRS reveals decreased NAA and increased choline and myoinositol compatible with axonal loss, dysmyelination, and gliosis. A prominent signal at 8.3 ppm in gray-and white-matter brain regions has been described in patients with adenylosuccinate lyase deficiency (Henneke et al., 2010). In patients with MSUD and acute metabolic decompensation, MRS demonstrates decreased NAA, methyl resonance of branched chain amino acids at 0.9 ppm, and elevated lactate (Cakmakci et al., 2009). In patients with X-linked adrenoleukodystrophy, the NAA/choline ratio (at a cutoff point of 0.5) has been shown to predict disease progression in six cases with cerebral disease (Eichler et al., 2002). Multislice proton MRS imaging was useful in identifying impending or beginning degeneration in white matter that still appeared normal on conventional MRI.
Imminent Death Prior to Diagnosis in a Child with a Suspected Inborn Error of Metabolism
Obtain samples needed for diagnosis when a child develops acute fatal metabolic decompensation and in cases of sudden and unexpected death (Olpin, 2004). A correct diagnosis may help families as they cope with their loss and enables appropriate counseling and prenatal diagnosis for subsequent pregnancies. Freeze plasma (separated from whole blood) and urine (CDC, 2003). Blood and bile (obtained by direct puncture of the gall bladder) specimens can also be collected, spotted on filter paper (two circles for each, about 25 µL), and dried before being sent to the laboratory. Obtain a skin sample under sterile technique (use alcohol swabs and not iodine, which interferes with cell growth), and store it at room temperature in tissue culture medium. When suspecting a storage disorder, obtain a small snip of skin and place it in glutaraldehyde for subsequent electron microscopic studies.
Management Considerations
The appropriate management of IEM depends upon the particular metabolic and/or molecular derangement. Therapeutic strategies may include one or more of the following approaches: (1) substrate reduction by dietary manipulation or precursor synthesis inhibition, (2) removal (or enhanced clearance) of the toxic metabolites, (3) replenishment of depleted metabolites or cofactor supplementation (or both), (4) enzyme (replacement or enhancement) therapy, and (5) cellular replacement (e.g., bone marrow, liver, heart, or kidney transplantation) (Chakrapani and Wraith, 2002; Leonard, 2004; Wilcken, 2003). Previous active exploration of gene therapy ended with the unexpected death of a study subject with ornithine transcarbamylase (OTC) deficiency. Reexamination of this approach led to demands for greater caution in future clinical trials (Hsich et al., 2002; Sands and Davidson, 2006). Meanwhile, gene therapy experiments are ongoing in various animal (mouse) models of disease (Koeberl et al., 2009). Other approaches under consideration are liver repopulation (hepatocyte infusion), chaperone-mediated therapy for diseases associated with residual enzyme activity, and the transplantation of stem cells with directed differentiation along specific lines (Desnick, 2004; Grompe, 2002; Kawashita et al., 2005). The rationale for the use of pharmacological chaperones is the demonstration that certain small molecules may rescue misfolded proteins in the endoplasmic reticulum and restore functional conformation (Gregersen et al., 2001; Pastores and Sathe, 2006). Certain drugs have also been shown in vitro to have the potential of reading through stop codons and may induce the synthesis of a full-length protein. A short-term study of gentamicin in a patient with McArdle disease and the R50X mutation failed to show modification of muscle metabolism (Schroers et al., 2006); these observations may be influenced by the short duration of the study or underlying gene defect. Clearly, more studies are required to examine the potential of this approach. Bezafibrate has been shown to increase carnitine palmitoyl transferase II (CPT2) messenger ribonucleic acid (mRNA), with normalization of enzyme activity in CPT2-deficient cultured fibroblasts and myoblasts. In a pilot trial including six patients treated with bezafibrate, FAO concentrations normalized in all patients, palmitoyl-l-carnitine oxidation increased, episodes of rhabdomyolysis were reduced, and quality of life improved without adverse effects (Bonnefont et al., 2009). These advances have changed the attitude about treatment for patients with IEM away from nihilism and hopelessness toward cautious optimism.
Special diets require attention to caloric needs and balanced nutrition; this is particularly true for minerals and supplements. Diseases managed with dietary restriction include the aminoacidopathies: phenylketonuria (PKU), MSUD, and homocystinuria. Recent clinical studies suggest that treatment of PKU patients with sapropterin (a cofactor of phenylalanine hydroxylase) provides better disease control and increases dietary phenylalanine tolerance, allowing significant relaxation, or even discontinuation, of dietary phenylalanine restriction (Blau et al., 2009). In classic Refsum disease, reduction in dietary phytanate results in normalization of the biochemical and clinical phenotype.
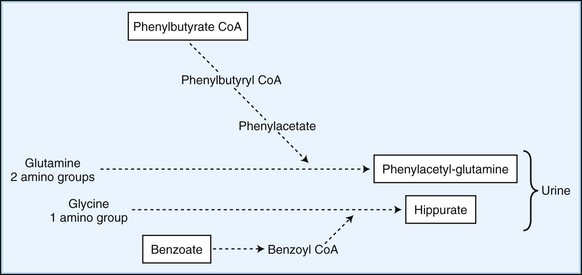
Employ methods to enhance the excretion or detoxification of toxic metabolites when dietary manipulation fails to correct their accumulation. In patients with hyperammonemia, give sodium benzoate and sodium phenylbutyrate, which conjugate with glycine and glutamine to facilitate nitrogen excretion (Fig. 62.1). In IVA, treatment with oral glycine (500 mg/kg) conjugates the highly toxic isovaleric acid to form a harmless byproduct excreted in the urine. Administration of cysteamine to patients with cystinosis promotes the formation of cysteine, which is subsequently excreted in the urine. Carnitine supplementation given to patients with organic acidemia prevents carnitine deficiency secondary to the formation and renal excretion of acylcarnitine compounds. During acute metabolic decompensation, dialysis and hemofiltration facilitates the rapid clearance of toxic metabolites. These techniques are already a part of the treatment of MSUD, carbamoylphosphate synthetase (CPS) deficiency, and hyperammonemic or leucine encephalopathy (Daschner and Schaefer, 2002). In patients with urea cycle defects, hemodialysis should be considered at ammonia levels above 600 mol/L (1000 g/L). Administration of glycerol trioleate and glycerol trierucate (Lorenzo oil) to asymptomatic patients with X-linked ALD may modify the disease course (Moser et al., 2005).
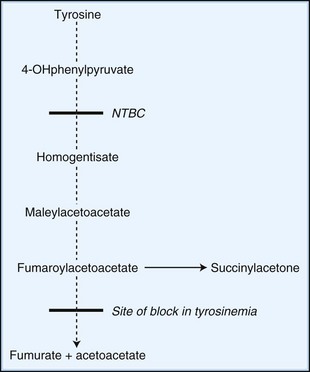
Novel therapeutic strategies for IEM use substrate synthesis inhibitors to block the production of toxic metabolites. In tyrosinemia type I (fumaroylacetoacetate hydrolase deficiency), NTBC (2-nitro-4-trifluoro-methylbenzoyl-1,3-cyclohexanedione) reduces the production of downstream metabolites of tyrosine degradation by inhibiting the enzyme, 4-hydroxyphenylpyruvate dioxygenase. This enzyme is involved in a reaction preceding the block (Fig. 62.2). In one study of NTBC in more than 300 patients with tyrosinemia type 1, 95% showed improvement of hepatic and kidney function (Grompe, 2001). In Gaucher disease type I, NB-DNJ (N-butyldeoxynojirimycin), miglustat administration leads to decreased liver and spleen volumes and a gradual but significant improvement in hematological parameters, with a decline in the levels of disease activity markers (Cox et al., 2000). NB-DNJ inhibits ceramide-specific glucosyltransferase, the first enzymatic step in glycosphingolipid (GSL) biosynthesis. Limiting substrate accumulation to a level that is sufficiently catabolizable by a mutant but partially active enzyme achieves metabolic homeostasis. Given this mechanism of action, miglustat may be potentially useful for other disorders of GSL metabolism (e.g., late-onset Tay-Sachs, Sandhoff disease, GM1-gangliosidosis, and Niemann-Pick disease type C [NPC]) (Pastores and Barnett, 2005).
Unfortunately, the miglustat trial in patients with either late-onset Tay-Sachs or Gaucher disease type 3 on enzyme replacement therapy was unable to prove any measurable benefit (Schiffmann et al., 2008; Shapiro et al., 2009). In patients with NPC, treatment with miglustat resulted in improvements in saccadic eye movements and swallowing, but impact on survival and eventual neurological prognosis appeared limited (Patterson et al., 2007).
Replenishing depleted substrates may partially correct the underlying defect in some IEM. In carnitine transport defects, the use of carnitine results in resolution of cardiomyopathy and prevention of further episodes of hypoketotic hypoglycemia. In other disorders, the production or binding affinity of a cofactor required for enzyme activity is impaired. Administration of pharmacological doses of the required supplement corrects the defect. Biotin, in some cases of PA and when given to children with biotinidase or holocarboxylase deficiency, leads to good clinical outcomes, except in the most severe forms with neonatal onset (Wolf, 2002). Vitamin B12 given for late-onset forms of MMA due to defects of adenosylcobalamin metabolism leads to a sustained decrease of toxic metabolites and a favorable developmental prognosis. Suppression of gut microbial propionate production (using metronidazole to inhibit anaerobic colonic flora) and dietary protein restriction are complementary approaches provided to children with MMA.
Additional examples of pharmacological correction include the use of tetrahydrobiopterin (BH4) to treat disorders of biopterin synthesis, rare variants of the hyperphenylalaninemia syndrome (Lambruschini et al., 2005). Although BH4 reduces elevated plasma phenylalanine levels by its action on liver phenylalanine hydroxylase, penetration of the blood-brain barrier is minimal. Only those with a peripheral-type defect respond. Tetrahydrofolate prevents demyelination in children with folate deficiency and dihydropteridine reductase deficiency. Assess the responsiveness to cofactor administration by controlled enzyme assays involving the patient’s cultured skin fibroblasts. Table 62.9 lists the cofactors used in various metabolic disorders and their recommended dosages.
Table 62.9 Cofactors Used in the Management of Various Inborn Errors of Metabolism
| Cofactor | Dose (mg/day) | Disorder |
|---|---|---|
| Betaine | 150* | Homocystinuria |
| MTHFR deficiency | ||
| Biotin | 10-20 | Propionic aciduria |
| Multiple carboxylase deficiency | ||
| Hyperlactacidemia due to pyruvate carboxylase deficiency | ||
| Carnitine | 50-100 PO | Branched-chain organic aciduria (MMA, PA, IVA) |
| 400 IV | Primary hyperammonemia | |
| Hyperlactacidemia | ||
| Fatty acid oxidation defects | ||
| Cobalamin (B12) | 1-2 | Methylmalonic aciduria |
| Folinic acid | 10-40 | Folinic-responsive seizures |
| Lorenzo oil | Asymptomatic X-linked (glycerol adrenoleukodystrophy trioleate and trierucate) | |
| Pyridoxine (B6) | 50-100 | Pyridoxine-responsive seizures, hyperoxaluria type 1, aromatic l-amino acid decarboxylase |
| Glutaric aciduria, homocystinemia | ||
| Riboflavin (B2) | 20-40 | Fatty acid oxidation defects† |
| Multiple acyl-CoA dehydrogenase deficiency | ||
| Thiamine (B1) | 10-50 | Maple syrup urine disease |
| Hyperlactacidemia due to pyruvate dehydrogenase deficiency |
IVA, Isovaleric acidemia; MMA, methylmalonic acidemia; MTHFR, methylene tetrahydrofolate reductase; PA, propionic acidemia.
* Dose is up to 150 mg/kg/day.
† Avoidance of fasting is critical; in emergency cases, where anorexia or vomiting precludes oral intake institute intravenous glucose administration.
Enzyme replacement therapy (ERT) reverses the hematological and visceral manifestations of Gaucher disease and may prevent or stabilize disease-related bone complications. This approach is also a consideration for the treatment of LSD due to single enzyme deficiencies. The relevant enzymes, produced from genetically manipulated mammalian cells in culture and subsequently modified, expose the appropriate sugar residues to facilitate targeted cell uptake. Once purified, management requires regular intravenous infusions of the recombinant enzyme. Beneficial effects have been noted in patients with Fabry disease, mucopolysaccharidosis (MPS) type I (Hurler-Scheie syndrome), MPS II (the mild variant of Hunter syndrome), MPS type VI (Maroteaux-Lamy syndrome), and GSD II (Pompe disease) (Pastores and Barnett, 2005). Enzyme therapy is also a consideration for other LSDs including Niemann-Pick disease.
Metabolic correction through cellular replacement by bone marrow transplantation (BMT)/hematopoietic stem cell transplantation (HSCT) has been performed in patients with LSD (e.g., MPS I, Gaucher disease type III) (Peters et al., 2003). In X-linked adrenoleukodystrophy, BMT has resulted in prolonged remission with reversal of MRI abnormalities and stabilization or improvement of motor function (Moser, 2006; Peters et al., 2004). Although BMT has altered the natural course of these diseases, donor limitation issues, procedural risks, and long-term care considerations (e.g., immunosuppression) exist. Advances in the methods of conditioning (e.g., nonmyeloablative procedures) prior to BMT and utilization of umbilical cord blood have addressed some of these concerns. Patients with IEM who may be suitable candidates for BMT should have serial follow-up visits incorporating neuropsychological and neuroradiological studies to time interventions prior to significant disease progression. This allows an outcome with minimal neurological sequelae. BMT is not appropriate for disorders characterized by rapid neurodegeneration, such as MPS II (severe Hunter syndrome) and MPS III (Sanfilippo syndrome).
Organ transplantation may be appropriate for disorders in which the metabolic defect is confined to the liver (e.g., Crigler-Najjar syndrome, hyperoxaluria type I) or leads to single organ failure (e.g., end-stage renal insufficiency in Fabry disease and hyperoxaluria type I, and liver failure in OTC deficiency, tyrosinemia, and GSD-IV) (Dhawan et al., 2005; Inderbitzin et al., 2005). In patients with tyrosinemia who do not respond to NTBC therapy or have evidence of hepatic malignancy, orthotopic liver transplantation is the treatment of choice. Microchimerism (the migration of donor-derived cells from the allograft) has occurred in a few patients following liver transplantation. However, this phenomenon is probably not sufficient to correct the systemic metabolic defect.
In addition to dealing with the medical problems of an affected child, provide genetic counseling. The inheritance of approximately 90% of IEMs is autosomal recessive. Of the remaining 10%, approximately two-thirds are X-linked and one-third autosomal dominant traits. Prenatal diagnosis is available for most IEM. A study that looked at reproductive decisions made by parents of children with IEM noted that 56% were receptive to future prenatal diagnosis, and 41% would choose to take measures to prevent another affected pregnancy (Read, 2002). The study also found that parents of children with IEM had higher scores on a stress index, lower scores on an adaptive behavior scale, and fewer persons in their social support network. Furthermore, the parents expressed greater worry about their child’s future and perceived difficulty in meeting the child’s extra care needs. These observations underscore the importance of early intervention, supportive care, and appropriate genetic counseling.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


