Inherited Metabolic Diseases of the Nervous System
John H. Menkes
William R. Wilcox
The diseases considered in this chapter result from a single mutant gene that codes for an enzymatic protein that in most instances is involved in a catabolic pathway. The consequent homeostatic disturbances produce a neurologic or developmental abnormality. The separation between conditions considered in this chapter and those in Chapter 3 is admittedly arbitrary. Both chapters deal with single-gene defects, except that for diseases covered in this chapter, the defective gene is normally expressed in one or more organs, not necessarily in the nervous system, and chemical analyses of tissues are frequently diagnostic. Conditions considered in Chapter 3 are also the result of a single defective gene, but one that is mainly or exclusively expressed in the nervous system. Consequently, these entities lack characteristic chemical abnormalities of tissues or body fluids.
Since 1975, almost all of the nearly 500 neurologic and neuromuscular diseases caused by known enzymatic or protein defects have been mapped to a specific chromosome region, and a large proportion of them have been cloned. In the course of these advances, a considerable amount of metabolic, molecular, and genetic detail has become available, the full discussion of which is outside the domain of this text. Instead, the emphasis of this chapter is on the clinical presentation of the diseases, their diagnosis and treatment, and, when known, the mechanisms that induce the neurologic deficits. For a more extensive discussion of the genetic and molecular basis of the neurologic disorders, the reader is referred to a text by Rosenberg and coworkers (1) and the compendium edited by Scriver and coworkers (2). In addition, we recommend two small paperback handbooks written by Clarke (3) and Hoffman and coworkers (3a) that are intended to make inborn errors of metabolism accessible to physicians who do not have an in-depth knowledge of biochemistry and molecular biology. For readers who are computer minded there is a helpful Web site at http://www.geneclinics.org.
Screening of newborns by tandem mass spectroscopy of amino acids and acylcarnitines has found an incidence of inborn errors of metabolism (excluding phenylketonuria) of approximately 15.7 in 100,000 live births (3b). These disorders are therefore individually relatively uncommon in the practice of pediatric neurology. Their importance rests, however, on the insight these disorders offer into the relationship between a genetic mutation, the resultant disturbance in homeostasis, and a disorder of the nervous system.
The mechanisms by which inborn errors of metabolism produce brain dysfunction remain largely uncertain, although, for some conditions, a plausible theory of pathogenesis has been proposed. Not all enzyme defects lead to disease; a large number of harmless metabolic variants exist. They include pentosuria, one of the original four inborn errors of metabolism described by Garrod (4), and several others listed in Table 1.1. Many of these variants were discovered when individuals in institutions for the mentally retarded were screened for metabolic defects at a time when their incidence in normal populations had not been determined. Over the last decade the imperfect relationship between the gene defect and its phenotypic expression has become evident. A single gene defect can result in a variety of phenotypic expressions, and, conversely, what has been assumed to be a single neurologic phenotype can have multiple genetic etiologies. Even for phenylketonuria, long considered to be the epitome of a metabolic disorder, the relationship between the mutation in the gene for phenylalanine hydroxylase, blood phenylalanine levels, and the ultimate result, impairment in cognitive function, is complex and unpredictable (5,5a).
An introduction to the fundamentals of molecular genetics is far beyond the scope of this text. The reader interested in this subject is referred to books by Strachan and Read (6), Lodish and colleagues (7), and Lewin (8).
For practical purposes, we will divide metabolic disorders into the following groups:
Disorders of amino acid metabolism
Disorders of renal amino acid transport
TABLE 1.1 Inborn Errors of Amino Acid Metabolism Without Known Clinical Consequences
Histidinemia (OMIM 235800)
Cystathioninemia (OMIM 219500)
Hyperprolinemia I (OMIM 239500)
Hyperprolinemia II (OMIM 239510)
Hydroxyprolinemia (OMIM 237000)
Hyperlysinemia (OMIM 238700)
Saccharopinuria (OMIM 247900)
Dibasic aminoaciduria I (OMIM 222690)
Alpha-aminoadipic aciduria (OMIM 204750)
Alpha-ketoadipic aciduria (OMIM 245130)
Sarcosinemia (OMIM 268900)
Disorders of carbohydrate metabolism and transport
Organic acidurias
Disorders of fatty acid oxidation
Lysosomal disorders
Disorders of lipid and lipoprotein metabolism
Peroxisomal disorders
Carbohydrate-deficient protein syndromes
Familial myoclonus epilepsies
Ceroid lipofuscinosis and other lipidoses
Disorders of serum lipoproteins
Disorders of metal metabolism
Disorders of purine and pyrimidine metabolism
Porphyrias
EVALUATION OF THE PATIENT SUSPECTED OF HAVING A METABOLIC DISORDER
The spectacular advances of molecular biology have facilitated the diagnosis and prevention of genetic diseases and have brought humanity to the threshold of gene therapy. The clinician must, therefore, strive for an early diagnosis of inborn errors of metabolism to offer treatment whenever possible, provide appropriate genetic counseling, and, in many instances, give parents an opportunity for an antenatal diagnosis on the occasion of a subsequent pregnancy (9,10).
Since the initial descriptions of phenylketonuria and maple syrup urine disease, the protean clinical picture of the various inborn metabolic errors has become apparent. As a consequence, these disorders must be included in the differential diagnosis of neurologic problems whenever other causes are not evident from the child medical history and physical examination.
Two questions should be considered: What type of patient should be suspected of having an inborn error of metabolism, and what tests should be included in the diagnostic evaluation? It is clear that the greater the suspicion of a metabolic disorder, the more intense the investigative process must be.
TABLE 1.2 Clinical Syndromes Suggestive of An Underlying Metabolic Cause | |
|---|---|
|
Table 1.2 lists some clinical syndromes (ranked by frequency) suggestive of an underlying metabolic cause.
A carefully obtained history and physical examination provide important clues to the presence of a metabolic disorder and its specific etiology (Table 1.3).
Metabolic investigations are less imperative for children who have focal neurologic disorders or who suffer from mental retardation in conjunction with major congenital anomalies. Dysmorphic features, however, have been found in some of the peroxisomal disorders, including Zellweger syndrome, in pyruvate dehydrogenase deficiency, in glutaric acidemia type II, and in Smith-Lemli-Opitz syndrome (11). These are given in Table 1.4.
When embarking on a metabolic investigation, procedures should be performed in ascending order of complexity and discomfort to patient and family.
Metabolic Screening
Routine screening of plasma and urine detects the overwhelming majority of disorders of amino acid metabolism and disorders manifested by an abnormality of organic acids as well as the common disorders of carbohydrate metabolism. Amino acid analysis can be performed by ion exchange chromatography, high-performance liquid chromatography, gas chromatography-mass spectrometry, or tandem mass spectrometry–mass spectrometry (TMS-MS). Analysis of organic acids is generally performed by gas–liquid chromatography with or without mass spectrometry. At our institution, metabolic screening usually includes plasma amino acids and urinary organic acids. The yield on these tests is low, and a high frequency of nonspecific or nondiagnostic abnormalities occurs. Urea cycle disorders are characterized by elevated concentrations of blood ammonia with the patient in the fasting state or on a high-protein intake (4 g/kg per day).
A number of genetic disorders are characterized by intermittent acidosis. Elevated lactic and pyruvic acid levels in serum or cerebrospinal fluid (CSF) are found in a number of the mitochondrial disorders. The lactate/pyruvate ratio may suggest the type of disorder. A normal ratio in
the face of an elevated lactate points to a disorder of pyruvate metabolism, whereas an elevated lactate/pyruvate ratio suggests a defect in nicotinamide adenine dinucleotide (NADH) oxidation, such as occurs in genetic defects of the mitochondrial electron transport chain. Measurements of lactate and pyruvate are prone to errors due to poor venipuncture technique and delayed sample handling, and lactate/pyruvate ratios should therefore be interpreted with caution. As a rule, measurements of arterial blood or CSF lactate and pyruvate are more reliable. In some mitochondrial disorders elevation of lactate cannot be documented from assays of body fluids but only by finding an elevation of lactate in brain by magnetic resonance spectroscopy (MRS).
the face of an elevated lactate points to a disorder of pyruvate metabolism, whereas an elevated lactate/pyruvate ratio suggests a defect in nicotinamide adenine dinucleotide (NADH) oxidation, such as occurs in genetic defects of the mitochondrial electron transport chain. Measurements of lactate and pyruvate are prone to errors due to poor venipuncture technique and delayed sample handling, and lactate/pyruvate ratios should therefore be interpreted with caution. As a rule, measurements of arterial blood or CSF lactate and pyruvate are more reliable. In some mitochondrial disorders elevation of lactate cannot be documented from assays of body fluids but only by finding an elevation of lactate in brain by magnetic resonance spectroscopy (MRS).
TABLE 1.3 Clinical Clues to the Diagnosis of Metabolic Diseases of the Nervous System | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 1.4 Abnormal Brain Development in Inborn Errors of Metabolism | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Determination of fasting blood sugar is also indicated as part of a metabolic workup. Should the suspicion for a metabolic disorder be high, any of these assays should be repeated when the child is ill with an intercurrent illness. For many disorders this is a safer approach than challenging the child with a protein or carbohydrate load. Other biochemical analyses required in the evaluation of a patient with a suspected metabolic defect include serum and urine uric acid, serum cholesterol, serum carnitine levels (including total, acyl, and free carnitine), immunoglobulins, thyroxine (T4), triiodothyronine (T3), serum copper, ceruloplasmin, magnesium, transferrin isoelectric focusing, and CSF glucose, protein, lactate, and amino acids. The assay of very long chain fatty acids is helpful in the diagnoses of adrenoleukodystrophy and other peroxisomal disorders.
Radiography
Radiographic examination of the vertebrae and long bones is helpful in the diagnosis of mucopolysaccharidoses, Gaucher diseases, Niemann-Pick diseases, and GM1 gangliosidosis. Neuroimaging studies, including magnetic resonance imaging (MRI), have been less helpful in pointing to any inborn metabolic error. Abnormalities such as agenesis of the corpus callosum and a large operculum seen in a few of the conditions covered in this chapter are nonspecific. The diagnostic yield of MRI is much higher in a child with mental retardation with or without seizures. As a rule, the more severe the mental retardation, the higher is the yield. Computed tomographic (CT) scanning contributes to the diagnosis of developmental delay in about 30% of children, whereas MRI shows a significant abnormality in 48% to 65% of patients presenting with global developmental delay (10). As is noted in Chapter 3 in the section entitled Diseases with Degeneration Primarily Affecting White Matter, MRI is also useful in the diagnosis of the various leukodystrophies. MRS is finding utility in diagnosing mitochondrial disorders and disorders of creatine synthesis.
TABLE 1.5 Inherited Metabolic Diseases Best Diagnosed from Analysis of Cerebrospinal Fluid | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Serum Lysosomal Enzyme Screen
Should the clinical presentation suggest a lysosomal disorder, a leukocyte lysosomal enzyme screen should be performed. In particular, assays for α-galactosidase, arylsulfatase, and the hexosaminidases can be run accurately in a number of centers. Occasionally, healthy individuals have been found with marked deficiency in one or another of the lysosomal enzymes. This “pseudodeficiency” is due to a mutation that alters the ability of the enzyme to degrade the artificial substrate used for the enzyme assay but does not impair enzymatic activity toward the natural substrate.
Cerebrospinal Fluid Analyses
In addition to the mitochondrial disorders, several other inherited metabolic diseases require analyses of cerebrospinal fluid for diagnosis. These are listed in Table 1.5.
Structural and Biochemical Alterations
In a number of metabolic disorders, notably the lipidoses and white matter degenerations, diagnosis requires clinical evaluation and combined microscopic, ultrastructural, and biochemical studies on biopsied tissue. In the past, a brain biopsy was required for this purpose, but neuroimaging studies have made this procedure unnecessary in almost all instances. It has become clear that, if adequately sought for structural and biochemical alterations can be detected outside the central nervous system (CNS) in almost every leukodystrophy or lipidosis. Table 1.6 shows what diseases (discussed in this chapter and Chapter 3) are likely to be diagnosed by examination of various tissues.
TABLE 1.6 Common Diseases Diagnosed by Study of Tissues | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
At this time, rectal biopsy and biopsies of other tissues such as kidney or tonsils are indicated only rarely. A liver biopsy is occasionally required to verify the actual enzymatic defect and is used to measure copper content in the diagnosis of Wilson disease, Menkes disease, and their variants. A conjunctival biopsy can be performed under local anesthetic and can be useful for excluding a lysosomal storage disease. When tissue biopsy suggests a specific metabolic disorder, highly sophisticated enzyme assays are required to confirm the diagnosis. Web sites that direct physicians to the various clinics that perform a given genetic test are maintained at http://www.genetests.org/ and http://www.biochemgen.ucsd.edu.
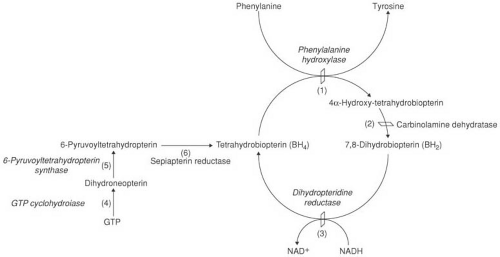 FIGURE 1.1. Phenylalanine metabolism. Phenylalanine is converted to tyrosine by the holoenzyme, phenylalanine hydroxylase (PAH). Phenylalanine hydroxylase requires tetrahydrobiopterin (BH4) as an active cofactor. BH4 is recycled by the sequential actions of (2) carbinolamine dehydratase and (3) dihydropteridine reductase. BH4 is synthesized in vivo from guanosine triphosphate (GTP) by a series of steps that involve (4) GTP cyclohydrolase and (5) 6-pyruvoyltetrahydropteridin (6-PT) synthase and (6) sepiatpterin reductase. Genetic defects in all these steps can result in hyperphenylalaninemia. A defect in GTP cyclohydrolase is seen also in dopa-responsive dystonia (see Chapter 3). (From Wilcox WR, Cedarbaum S. Amino acid metabolism. In: Rimoin DL, Connor JM, Pyeritz RE, et al., eds. Principles and practice of medical genetics, 4th ed. New York: Churchill Livingstone, 2002;2405–2440.) |
DISORDERS OF AMINO ACID METABOLISM
Phenylketonuria (Online Mendelian Inheritance in Man [OMIM] Database, Number 261600)
Phenylketonuria (PKU) has long been the prototype for an inborn metabolic error that produces serious neurologic symptoms. We will therefore consider it in more detail than its frequency would warrant. Fölling, in 1934, first called attention to the condition in a report of 10 mentally defective patients who excreted large amounts of phenylpyruvic acid (12). The disease has since been found in all parts of the world, although it is rare in blacks and Ashkenazi Jews. Its incidence in the general population of the United States, as determined by screening programs, is approximately 1 in 14,000 (13).
Molecular Genetics and Biochemical Pathology
Phenylketonuria is an autosomal recessive disorder that results from a mutation in the gene that codes for phenylalanine hydroxylase (PAH), the enzyme that hydroxylates phenylalanine to tyrosine. The complete hydroxylation system consists in part of PAH, a PAH stabilizer, the tetrahydrobiopterin cofactor (BH4), dihydropteridine reductase (DHPR), which is required to recycle BH4, and a BH4-synthesizing system. This system involves guanosine triphosphate (GTP) cyclohydrolase and 6-pyruvoyl tetrahydropteridine synthetase (Fig. 1.1) (14).
PAH is normally found in liver, kidney, and pancreas but not in brain or skin fibroblasts. The enzyme is an iron-containing metalloprotein dimer of identical subunits with a molecular weight of approximately 100,000. The gene coding for the enzyme has been cloned and localized to the long arm of chromosome 12 (12q22–q24). The gene is approximately 90 kilobases (kb) long with 13 exons, and codes for a mature RNA of approximately 2,400 bases. Availability of this clone has facilitated the molecular genetic analysis of patients with PKU and has confirmed that PKU is the consequence of numerous mutant alleles arising in various ethnic groups. The majority of these mutations result in deficient enzyme activity and cause hyperphenylalaninemia. Mutations occur in all 13 exons of the gene and flanking sequence. Some cause phenylketonuria; others cause non-PKU hyperphenylalaninemia, whereas still others are silent polymorphisms present on both
normal and mutant chromosomes (15,16). Some PAH alleles are more prevalent; five account for approximately 60% of European mutations and they tend to cluster in regions or are on only one of a few haplotypes. More than 495 mutations have been recorded. They are catalogued at http://www.pahdb.mcgill.ca.
normal and mutant chromosomes (15,16). Some PAH alleles are more prevalent; five account for approximately 60% of European mutations and they tend to cluster in regions or are on only one of a few haplotypes. More than 495 mutations have been recorded. They are catalogued at http://www.pahdb.mcgill.ca.
Most mutations are missense mutations, although splice, nonsense, and silent mutations as well as single–base-pair frameshifts and larger deletions and insertions have been found. Most patients with PKU are compound heterozygotes rather than homozygotes in the precise meaning of the term (17). As a rule, the biochemical phenotype (i.e., the degree of phenylalanine elevation) does not correlate with the activity of PAH as predicted from the genetic mutation. In addition, no good correlation exists between PAH activity and intellectual function of untreated patients (18). Scriver (3,5) and others have pointed out that various environmental factors and modifying genes, such as the blood–brain barrier phenylalanine transporters, and variations in brain phenylalanine consumption rate may affect intellectual function in phenylketonuric patients (19,20).
The infant with classic PKU is born with only slightly elevated phenylalanine blood levels, but because of the absence of PAH activity, the amino acid derived from food proteins and postnatal catabolism accumulates in serum, CSF, and brain and is excreted in large quantities. In lieu of the normal degradative pathway, phenylalanine is converted to phenylpyruvic acid, phenylacetic acid, and phenylacetylglutamine.
The transamination of phenylalanine to phenylpyruvic acid is sometimes deficient for the first few days of life, and the age at which phenylpyruvic acid can be first detected varies from 2 to 34 days. From the first week of life, o-hydroxyphenylacetic acid also is excreted in large amounts.
In addition to the disruption of phenylalanine metabolism, tryptophan and tyrosine are handled abnormally. Normally, transport across the blood–brain barrier of a large neutral amino acid such as phenylalanine is determined by its plasma concentration and its affinity to the stereo-specific L-type amino acid carrier system (20). Supraphysiologic levels of phenylalanine competitively inhibit transport of tryptophan and tyrosine across the blood–brain barrier (21). Intestinal transport of L-tryptophan and tyrosine is also impaired in PKU, and fecal content of tryptophan and tyrosine is increased. These abnormalities are reversed by dietary correction of the plasma phenylalanine levels (22). Miyamoto and Fitzpatrick suggested that a similar interference might occur in the oxidation of tyrosine to 3-(3,4-dihydroxyphenyl) alanine (dopa), a melanin precursor, and might be responsible for the deficiency of hair and skin pigment in phenylketonuric individuals (23). The biochemical pathology of PKU is reviewed to a greater extent by Scriver and Kaufman (14).
Pathologic Anatomy
Alterations within the brain are nonspecific and diffuse. They involve gray and white matter, and they probably progress with age. Three types of alterations exist.
Interference with the Normal Maturation of the Brain
Brain growth is reduced, and microscopic examination shows impaired cortical layering, delayed outward migration of neuroblasts, and heterotopic gray matter (24). Additionally, the amount of Nissl granules is markedly deficient. This is particularly evident in those areas of the brain that are not fully developed at birth. Dendritic arborization and the number of synaptic spikes are reduced within the cortex (25). These changes point to a period of abnormal brain development extending from the last trimester of gestation into postnatal life (Fig. 1.2).
Defective Myelination
Defective myelination may be generalized or limited to areas where one would expect postnatal myelin deposition. Except for adult patients with PKU with neurologic deterioration, products of myelin degeneration are unusual (26). Myelin is usually relatively pale, and a mild degree of gliosis (Fig. 1.3) and irregular areas of vacuolation (status spongiosus) can be present. Areas of vacuolation usually are seen in central white matter of the cerebral hemispheres and in the cerebellum.
Diminished or Absent Pigmentation of the Substantia Nigra and Locus Ceruleus
Because substantia nigra and locus ceruleus are normally pigmented in albinos and tyrosinase activity cannot be demonstrated in normal neurons within the substantia nigra, diminished or absent pigmentation is not a result of tyrosinase inhibition by phenylalanine or its derivatives (27). Instead, neuromelanogenesis in the phenylketonuric patient must be interrupted at some other metabolic point, such as the metal-catalyzed pseudoperoxidation of dopamine derivatives probably responsible for the melanization of lipofuscin in the substantia nigra (26).
Clinical Manifestations
Phenylketonuric infants appear healthy at birth. In untreated infants, vomiting, which at times is projectile, and irritability are frequent during the first 2 months of life. By 4 to 9 months, delayed intellectual development becomes apparent (28). In the untreated classic case, mental retardation is severe, precluding speech and toilet training. Children in this category have an IQ below 50. Seizures, common in the more severely retarded, usually start before 18 months of age and can cease spontaneously. During
infancy, they often take the form of infantile spasms, later changing into tonic-clonic attacks.
infancy, they often take the form of infantile spasms, later changing into tonic-clonic attacks.
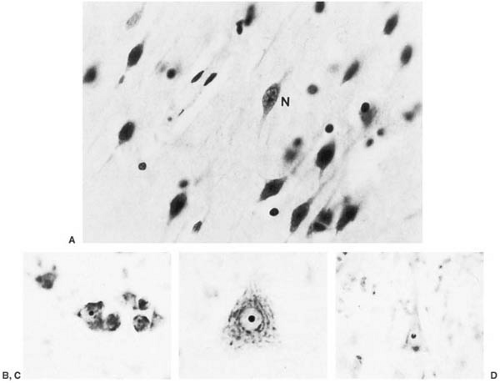 FIGURE 1.2. Phenylketonuria. A: Cresyl-violet-stained section showing spindle-shaped immature neuron (N) (×350). (Courtesy of Dr. Nathan Malamud, Langley Porter Neuropsychiatric Institute, San Francisco, CA.) B: Photomicrograph of Nissl-stained giant Betz cell from a healthy 4-month-old child. C: Photomicrograph of Nissl-stained Betz cells from a healthy 14-year-old child. D: Photomicrograph of Nissl-stained Betz cell from a 19-year-old with untreated phenylketonuria. Patient’s developmental level was 4.6 months at age 5 years and 10 months; he was microcephalic and had seizures commencing at 10 years of age. Note that the Betz cells are reduced in size, with a pale cytoplasm and few well-formed Nissl granules. These cytoarchitectural abnormalities are nonspecific. (B, C, and D from Bauman ML, Kemper TL. Morphologic and histoanatomic observations of the brain in untreated human phenylketonuria. Acta Neuropathol 1982;58:55–63. With permission.) |
The untreated phenylketonuric child is blond and blue-eyed, with normal and often pleasant features. The skin is rough and dry, sometimes with eczema. A peculiar musty odor, attributable to phenylacetic acid, can suggest the diagnosis. Significant neurologic abnormalities are rare, although hyperactivity and autistic features are not unusual. Microcephaly may be present as well as a mild increase in muscle tone, particularly in the lower extremities. A fine, irregular tremor of the outstretched hands is seen in approximately 30% of the patients. Parkinsonian-like extrapyramidal symptoms also have been encountered (29). The plantar response is often extensor.
A variety of electroencephalographic (EEG) abnormalities has been found, but hypsarrhythmic patterns, recorded even in the absence of seizures, and single and multiple foci of spike and polyspike discharges are the most common (30).
MRI is abnormal in almost every patient, regardless of when treatment was initiated. On T2-weighted imaging, one sees increased signal in the periventricular and subcortical white matter of the posterior hemispheres. Increased signal can extend to involve the deep white matter of the posterior hemispheres and the anterior hemispheres. No signal abnormalities are seen in brainstem, cerebellum, or cortex, although cortical atrophy may be present (Fig. 1.4) (31,32). The severity of the abnormality is unrelated to the patient’s IQ but is significantly associated with the phenylalanine level at the time of imaging. In adult patients with PKU who had come off their diets, resumption of dietary treatment can improve MRI abnormalities within
a few weeks or months, an observation that strongly suggests that at least some of the MRI changes are the result of edema (32).
a few weeks or months, an observation that strongly suggests that at least some of the MRI changes are the result of edema (32).
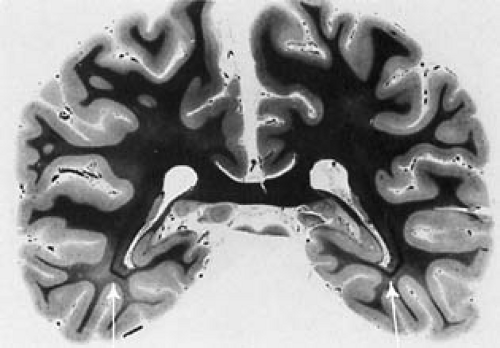 FIGURE 1.3. Phenylketonuria. Cerebrum of a 35-year-old man stained for myelin by the Loyez method. The individual was never treated. He sat at 1 year and walked at 3 years. He was microcephalic and spastic and never developed speech. Death was caused by pulmonary tuberculosis and dehydration. The visual radiations (arrows) stand out against the background of persisting pallor of the association tracts and nonspecific thalamic radiations. In a healthy brain of that age, the visual radiations are not distinguishable by tonality of staining from the completely myelinated white matter. (From Bauman ML, Kemper TL. Morphologic and histoanatomic observations of the brain in untreated human phenylketonuria. Acta Neuropathol 1982;58:55–63. With permission.) |
Heterozygous mothers tend to have elevated plasma phenylalanine levels and somewhat reduced tyrosine values during the latter part of pregnancy and after delivery. Mothers suffering from PKU have a high incidence of spontaneous abortions. It is now clear that when maternal blood phenylalanine levels are greater than 20 mg/dL (1,212 μmol/L) during pregnancy, fetal brain damage is almost inevitable, with mental retardation encountered in 92% of offspring and microcephaly in 73%. Offspring have an unusual facies: upturned nose, underdeveloped philtrum, and a thin upper lip (33). Additionally, a significant incidence of congenital heart disease and prenatal and postnatal growth retardation occurs (34). MRI in these children shows hypoplasia or partial agenesis of the corpus callosum and delayed myelination (35). These observations are best explained by a deleterious effect of elevated phenylalanine on the myelinating ability of oligodendrocytes.
Much, if not all, of the fetal damage appears to be preventable by placing phenylketonuric mothers on a phenylalanine-restricted diet before conception and maintaining blood phenylalanine levels below 6 mg/dL (360 μmol/L) throughout pregnancy (36). In the data compiled by Koch and coworkers the optimal outcome, as measured by the Wechsler Intelligence Scale for Children (WISC) score of offspring obtained at 7 years of age, was obtained when maternal blood phenylalanine levels of 120 to 360 μmol/L (2 to 6 mg/dL) were obtained by 8 to 10 weeks’ gestation and maintained at those levels throughout pregnancy. However, there was considerable scatter in IQ even when the phenylalanine levels were maintained below 5 mg/dL (37). Birth weight and head circumference in infants of mothers so managed were normal, and no evidence existed for fetal nutritional deficiency. Offspring of mothers who experience mild PKU as defined by phenylalanine levels of less than 400 μmol/L (6.6 mg/dL) appear to be normal, and mothers do not require dietary intervention (38).
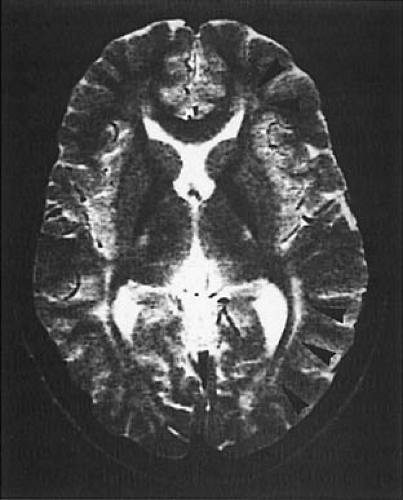 FIGURE 1.4. T2-weighted magnetic resonance imaging of a 16-year-old girl with phenylketonuria. The girl was treated from early infancy, and the diet was stopped at 12 years of age. At the time of the scan, her neurologic examination was normal, but her phenylalanine level was 27.0 mg/dL (1,639 μmol/L). The scan shows areas of high signal, particularly in the parieto-occipital areas, but also at the tips of the anterior horns of the lateral ventricles. (Courtesy of Dr. Alan J. Thompson, Neurorehabilitation Section, Institute of Neurology, University of London.) |
The pathogenesis of mental retardation in PKU is not completely understood (39). No evidence exists that phenylpyruvic acid or any of the other phenylalanine metabolites are neurotoxic at concentrations in which they are seen in PKU (40). Probably no single factor is responsible; instead, impairment of amino acid transport across the blood–brain barrier; disruption of the brain amino acid pool; with consequent defective proteolipid protein synthesis, impaired myelination, and low levels of neurotransmitters, such as serotonin, are responsible to varying degrees (41,42).
Diagnosis
With the worldwide practice of neonatal screening for phenylketonuria, the diagnosis is usually made during the first week of life, and it is now rare to encounter an older infant or child with undiagnosed PKU. As mentioned in the section on Molecular Genetics and Biochemical Pathology, plasma phenylalanine levels are elevated in the cord blood of phenylketonuric infants and increase rapidly within a few hours of birth. A screening program that involves spectrofluorometric or microbiologic estimation of blood phenylalanine is used commonly. Tandem mass spectrometric analysis of phenylalanine and other amino acids as well as of the acylcarnitines of the various organic acids is being used in some laboratories (43), and trials of this method for newborn screening are being instituted in several states. Whatever the method, the screening test requires a drop of whole blood placed on a filter paper. If the test is positive [a value greater than 2 to 4 mg/dL (120 to 240 μmol/L)], the diagnosis is confirmed by quantitative analysis of phenylalanine and tyrosine in blood. Although most infants whose blood phenylalanine levels are above the threshold value do not have PKU, such patients require prompt reevaluation by an appropriate laboratory to determine whether hyperphenylalaninemia is persistent and whether it is caused by PAH deficiency.
Of considerably greater concern are the false-negative test results. It is now clear that routine screening tests, when correctly performed after 12 hours of life, detect all infants with classic PKU. In a group of such infants studied by Koch and Friedman the lowest phenylalanine value recorded at 24 hours was 5.6 mg/dL (339 μmol/L); at 48 hours, 7.5 mg/dL (454 μmol/L); and at 72 hours, 8.4 mg/dL (509 μmol/L). Thus, even at 24 hours, none of the infants with classic PKU would have escaped detection (44). The Canadian experience of Hanley and coworkers is similar (45). As a rule, breast-fed PKU infants have higher phenylalanine levels than those who are formula fed. In some infants with mild hyperphenylalaninemia, the increase in blood phenylalanine is sufficiently slow to allow them to escape detection unless an assay is used, such as tandem mass spectroscopy, that can measure both phenylalanine and tyrosine, which decreases false-positive results. Some screening agencies obtain a second screening blood specimen from all infants whose first blood specimen was drawn during the first 24 hours of life (46,47).
Most infants with elevated blood phenylalanine detected by means of the newborn screening program do not have classic PKU; instead they have a transient or mild form of hyperphenylalaninemia that is usually benign. Patients with mild PKU have phenylalanine levels between 600 and 1200 μmol/L (10 to 20 mg/dL) on an unrestricted diet, and patients with mild hyperphenylalaninemia have levels below 600 μmol/L (10 mg/dl) on an unrestricted diet (48). A large proportion of these patients represent compound heterozygotes for a mutation that abolishes catalytic activity of PAH and one that reduces it (16,49,50,51). With normal protein intakes, the majority of such infants appear to have unimpaired intelligence, even when untreated (48).
Elevated blood phenylalanine levels also are observed in 25% of premature infants and occasionally in full-term newborns. In all of these patients, tyrosine levels are increased to a much greater extent than are phenylalanine levels.
Treatment
Two treatment strategies could be employed in PKU: modification of the phenotypic expression of the defective gene and definitive treatment by correcting the gene defect. Only the first approach has been used in clinical practice.
On referral of an infant with a confirmed positive screening test result, the first step is quantitative determination of serum phenylalanine and tyrosine levels. All infants whose blood phenylalanine concentration is greater than 10 mg/dL (600 μmol/L) and whose tyrosine concentration is low or normal (1 to 4 mg/dL) should be started on a low-phenylalanine diet immediately. Infants whose blood phenylalanine concentrations remain below 10 mg/dL (600 μmol/L) on an unrestricted diet are generally considered not to require a diet (54).
The accepted therapy for classic PKU is restriction of the dietary intake of phenylalanine by placing the infant on one of several low-phenylalanine formulas. The diet should be managed by a team consisting of a nutritionist, a physician with expertise in metabolic disorders, and a person to ensure dietary compliance. To avoid symptoms of phenylalanine deficiency, regular formula is added to the diet in amounts sufficient to maintain blood levels of the amino acid between 2 and 6 mg/dL (120 and 360 μmol/L). Generally, patients tolerate this diet quite well, and within 1 to 2 weeks, the serum concentration of phenylalanine becomes normal. Serum phenylalanine determinations are essential to ensure adequate regulation of diet. These are performed twice weekly during the first 6 months of life and twice monthly thereafter.
Strict dietary control should be maintained for as long as possible, and most centers strive to keep levels below 6.0 mg/dL (360 μmol/L), even in patients with moderate and mild PKU. Samples of low-phenylalanine menus are given by Buist and colleagues (55), and the nutritional problems inherent in prolonged use of a restricted diet are discussed by Acosta and colleagues (56). Dietary lapses are common, particularly in patients 15 years or older (57). They frequently are accompanied by progressive white
matter abnormalities on MRI. Some workers have suggested supplementation of the low-phenylalanine diet with tyrosine, but no statistical evidence exists that this regimen results in a better intellectual outcome. This possibly could be due to failure to achieve adequate tyrosine levels in the brain even with tyrosine supplementation of 300 mg/kg (58). Failure to treat patients with mild hyperphenylalaninemias does not appear to produce either intellectual deficits or MRI abnormalities.
matter abnormalities on MRI. Some workers have suggested supplementation of the low-phenylalanine diet with tyrosine, but no statistical evidence exists that this regimen results in a better intellectual outcome. This possibly could be due to failure to achieve adequate tyrosine levels in the brain even with tyrosine supplementation of 300 mg/kg (58). Failure to treat patients with mild hyperphenylalaninemias does not appear to produce either intellectual deficits or MRI abnormalities.
Patients with mild PKU or mild hyperphenylalaninemia have been treated successfully with tetrahydrobiopterin (BH4; 10 to 20 mg/kg per day) (59,59a). Most of these individuals have at least one missense mutation in the gene for phenylalanine hydroxylase, leading to some residual enzyme activity (60). The mechanism for this effect is unknown, as is the question of how to best select patients responsive to the cofactor. BH4 is being tested, but it is not yet approved for therapy in the United States.
Dietary therapy has for the greater part been effective in preventing mental retardation in patients with severe PKU. It has become apparent that the outcome depends on several factors. Most important is the age at which the diet was initiated. Smith and coworkers found that patients’ IQ fell approximately 4 points for each month between birth and start of treatment (61). The average phenylalanine concentration while receiving treatment also affects outcome, with optimal average phenylalanine levels in the most recent cohorts being 5.0 to 6.6 mg/dL (300 to 400 μmol/L). Additionally, hypophenylalaninemia during the first 2 years of life [i.e., the length of time that phenylalanine concentration was below 2.0 mg/dL (120 μmol/L)] also affected outcome adversely. Even patients with normal IQ scores and with the most favorable diagnostic and treatment characteristics have lower IQs than other members of their families and suffer from cognitive deficits, educational difficulties, and behavioral problems, notably hyperactivity (61,62,63). As was already noted, dietary supplementation with tyrosine, although used in several centers, does not appear to improve performance on neuropsychologic tests (64). The most likely explanations for these deficits are inadequate dietary control and unavoidable prenatal brain damage from elevated phenylalanine (see Fig. 1.2) (24).
When patients who already have developed symptoms caused by classic PKU are placed and maintained on a low-phenylalanine diet, the epilepsy comes under control and their EEGs normalize. Microcephaly, if present, can correct itself, and abnormally blond hair regains its natural color.
Considerable uncertainty exists about when, if ever, to terminate the diet (65). In the series of Waisbren and coworkers (66), one-third of youngsters whose diet was discontinued at 5 years of age had a reduction in IQ of 10 points or more during the ensuing 5 years. The blood phenylalanine level when the children were off the restrictive diet predicted the change in IQ. Of the children whose IQs dropped 20 or more points, 90% had blood phenylalanine levels of 18 mg/dL (1,090 μmol/L) or higher, and 40% of those whose IQs rose 10 points or more had a phenylalanine level of less than 18 mg/dL (1,090 μmol/L). In young adult patients with PKU, discontinuation of the diet was accompanied by progressive spasticity and worsening white matter abnormalities (67). Reinstitution of the diet results in clear clinical improvement and resolution of new MRI abnormalities. Reports such as these speak against early relaxation of the restrictions on phenylalanine intake and indicate that dietary therapy for patients with classical PKU should be life-long (68,69).
The relative inadequacies of dietary therapy underscore the need for a more definitive approach to the treatment of PKU. Allogeneic or autologous bone marrow transplants are being used for the treatment of a variety of genetic diseases (Table 1.7). The likelihood that this procedure will cure or at least stabilize a genetic disease depends on the tissue-specific expression of the normal gene product, the patient’s clinical symptoms, and the cellular transport of the normal gene product. In PKU, the defective enzyme is not normally expressed in bone marrow–derived cells, and bone marrow transplantation has no therapeutic value (see Table 1.7).
Another approach to treating the patient with PKU is the introduction of PAH gene into affected hepatic cells. Recombinant viruses containing human PAH have been introduced into mouse hepatoma cells, where they are able to continue expressing the human enzyme, lowering the phenylalanine level and normalizing coat color (70). The next step would be to find a virus that can infect human liver, maintain itself there without inducing damage to the organ, and allow the human gene to continue functioning in the new host. Ding and colleagues reviewed various approaches for nonviral gene transfer (71).
Other Conditions Characterized by Hyperphenylalaninemia
About 1% of infants with persistent hyperphenylalaninemia do not have PKU but lack adequate levels of BH4, the cofactor in the hydroxylation of phenylalanine to tyrosine. This can be due to a defect in BH4 biosynthesis or to a deficiency of dihydropteridine reductase (DHPR), one of the enzymes involved in the regeneration of BH4. These conditions are depicted in Table 1.8.
Dihydropteridine Reductase Deficiency (OMIM 231630)
The first and most common of these conditions to be recognized is characterized by undetectable DHPR activity in liver, brain, and cultured fibroblasts but normal hepatic PAH activity (72). DHPR is responsible for the regeneration
of BH4 from quinoid dihydrobiopterin (see Fig. 1.1). BH4 levels are low in blood, urine, CSF, and a number of tissues. Because BH4 is an essential coenzyme for hydroxylation of not only phenylalanine, but also tyrosine and tryptophan, affected children show a defect in the synthesis of dopamine, norepinephrine, epinephrine, and serotonin.
of BH4 from quinoid dihydrobiopterin (see Fig. 1.1). BH4 levels are low in blood, urine, CSF, and a number of tissues. Because BH4 is an essential coenzyme for hydroxylation of not only phenylalanine, but also tyrosine and tryptophan, affected children show a defect in the synthesis of dopamine, norepinephrine, epinephrine, and serotonin.
TABLE 1.7 Some Neurogenetic Diseases that can be Treated with Bone Marrow Transplantation (BMT) or Enzyme Replacement Therapy (ERT) | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The clinical picture of DHPR deficiency is one of developmental delay associated with the evolution of marked hypotonia, involuntary movements, oculogyric crises, and tonic-clonic and myoclonic seizures. None of these symptoms resolve with restriction of phenylalanine intake (73,74). Progressive intracranial calcifications can be demonstrated by CT scanning. These might be the consequence of reduced intracranial tetrahydrofolate (75) because folate deficiency, whether it is caused by inadequate intake or defective absorption, can induce intracranial calcifications. MRI demonstrates white matter abnormalities and cystic loss of parenchyma (76).
TABLE 1.8 Disorders of Phenylalanine Metabolism — the Hyperphenylalaninemias | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
Treatment for patients with DHPR deficiency requires restriction of phenylalanine intake and administration of catechol and serotonin precursors. The former is given as levodopa-carbidopa (Sinemet) and the latter as 5-hydroxytryptophan (8 to 10 mg/kg per day). Additionally, folinic acid (12.5 mg per day) is added to the diet (77). Treatment of the other variants is discussed by Shintaku (78).
6-Pyruvoyl-Tetrahydropterin (6-PT) Synthase Deficiency (OMIM 261640)
Increased phenylalanine levels can result from inadequate biopterin synthesis. In 6-pyruvoyl-tetrahydropterin (6-PT) synthase deficiency the defect is localized to the synthetic pathway of BH4 at the point of the formation of 6-PT (see Fig. 1.1). The enzyme deficiency can be complete, partial, or transient or might affect only nonneural tissue (73,79,80).
The clinical picture of this entity is much like that of DHPR deficiency: progressive neurologic deterioration highlighted by hypotonia, involuntary movements, and seizures. Diagnosis of this variant depends on normal assays for PAH and dihydropteridine reductase and on determination of urinary or CSF pterins (81). MRI in this disorder is similar to that seen in classical PKU (82).
GTP Cyclohydrolase Deficiency (OMIM 233910)
Another rare cause for persistent hyperphenylalaninemia is a defect of GTP-cyclohydrolase, needed for the first step of BH4 biosynthesis (see Fig. 1.1) (83,84). Symptoms include hypotonia, seizures, and hyperthermia. Mutations in GTP-cyclohydrolase are also responsible for dopa-responsive dystonia, a condition covered in Chapter 3 in the section on Primary Dystonia. Symptoms in this condition differ considerably from those seen in hyperphenylalaninemia. The best explanation for the phenotypic diversity is that the mutant enzyme has a dominant negative effect on the normal enzyme (85).
TABLE 1.9 Clinical and Genetic Features of the Tyrosinemias | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Sepiapterin Reductase Deficiency (OMIM 182125)
The clinical picture in this condition does not differ significantly from that of DHPR deficiency, namely dystonia, spasticity, and progressive mental retardation. Blood phenylalanine levels are normal, and the diagnosis depends on demonstrating elevated CSF sepiapterin, biopterin, and dihydrobiopterin levels (86,87).
Carbinol Dehydratase Deficiency (OMIM 126090)
Children with this condition present with mild hyperphenylalaninemia, and the diagnosis is made by finding elevated urinary 7-biopterin (88).
Tyrosinosis and Tyrosinemia
Maple Syrup Urine Disease (OMIM 248600)
Maple syrup urine disease (MSUD) is a familial cerebral degenerative disease caused by a defect in branched-chain amino acid metabolism and characterized by the passage of urine that has a sweet, maple syrup–like odor. It was first described in 1954 by Menkes and coworkers (101). Since then, numerous other cases have been diagnosed throughout the world, and its incidence is estimated at 1 in 220,000 births (102). In some inbred populations, such as the Mennonites, the incidence is as high as 1 in 176 births (103). The disease occurs in all races and is transmitted in an autosomal recessive manner.
Molecular Genetics and Biochemical Pathology
MSUD is characterized by the accumulation of three branched-chain ketoacids: α-ketoisocaproic acid, α-ketoisovaleric acid, and α-keto-β-methylvaleric acid, the derivatives of leucine, valine, and isoleucine, respectively (104,105). Their accumulation is the consequence of a defect in oxidative decarboxylation of branched-chain ketoacids (Fig. 1.5).
The branched-chain α-ketoacid dehydrogenase complex is located within the mitochondrial inner membrane matrix compartment. It is a multienzyme complex comprising six proteins: E1α and E1β. which form the decarboxylase; E2; E3; and a branched-chain–specific kinase and phosphatase. The last two enzymes regulate the activity of the complex by phosphorylating and dephosphorylating the dehydrogenase. E1 is a thiamin pyrophosphate-dependent enzyme. The second enzyme (E2), dihydrolipoyltransacylase, transfers the acyl group from the first enzyme to coenzyme A. The third enzyme (E3), dihydrolipoyldehydrogenase, a flavoprotein, reoxidizes the disulfhydryl form of lipoamide. The same enzyme is common to other α-ketoacid dehydrogenases, such as pyruvate and α-ketoglutarate dehydrogenase (106). The complex removes carboxyl groups from all three branched-chain ketoacids and converts those ketoacids to their respective coenzyme A derivatives (see Fig. 1.5, step 2). Chuang and coworkers reviewed the crystal structure of the various enzyme components and the structural basis for the various MSUD mutations (103).
With six genes involved in the function of the branched-chain ketoacid dehydrogenase complex, considerable room for heterogeneity exists. Mutations in the genes for E1α, E1β, E2, and E3 have been described, with many MSUD patients being compound heterozygotes. These induce a continuum of disease severity that ranges from the severe, classic form of MSUD to mild and intermittent forms.
As a consequence of the enzymatic defect, the branched-chain ketoacids accumulate in serum and CSF and are excreted in large quantities in urine (105). Plasma levels of the respective amino acids (e.g., leucine, isoleucine, and valine) are elevated secondary to the increase in ketoacid concentrations. Alloisoleucine, which is formed by transamination of α-keto-β-methylvaleric acid, also has been found in serum (107). In some cases, the branched-chain hydroxyacids, most prominently α-hydroxyisovaleric acid (108), are excreted also. Sotolone, a derivative of α–ketobutyric acid, the decarboxylation of which is impaired by accumulation of α-keto-β-methylvaleric acid, is responsible for the characteristic odor of the patient’s urine and perspiration (109).
Pathologic Anatomy
Structural alterations in the nervous system in untreated infants with MSUD are similar to, but more severe than, those seen in PKU. In infants dying during the acute phase of the disease, diffuse edema occurs (101). The cytoarchitecture of the cortex is generally immature, with fewer cortical layers and the persistence of ectopic foci of neuroblasts, an indication of disturbed neuronal migration. Dendritic development is abnormal, and the number of oligodendrocytes and the amount of myelin are less than would be seen in a healthy brain of comparable age (110). Marked astrocytic gliosis and generalized cystic degeneration occur (111). Little clinical or pathologic evidence exists for demyelination in patients who are treated early (112,113). On chemical examination, the concentration of myelin lipids is markedly reduced, with cerebrosides, sulfatides, and proteolipid protein almost completely absent. These abnormalities are not found in infants dying of the disease within the first days of life or in patients treated by restriction of branched-chain amino acid intake (113).
Clinical Manifestations
Various mutations result in five fairly distinct clinical phenotypes of MSUD. Patients can be homozygotes for the same allele or compound heterozygotes for different alleles. The classic form of MSUD accounts for approximately 75% of patients (103). Mutations in the E1 decarboxylase component of the enzyme are associated with this phenotype (114). In the original four patients reported by Menkes and coworkers (101) as well as in subsequent cases, dystonia, opisthotonos, intermittent increase in muscle tone, and respiratory irregularities appeared within the first week of life in babies apparently healthy at birth (115). Subsequently, rapid deterioration of the nervous system occurred, and all but one died within 1 month. In some patients, cerebral edema is marked and can be fatal (116). Other patients, spastic and intellectually retarded, survived without treatment for several years. A fluctuating ophthalmoplegia correlates in intensity with serum leucine levels (117). Presentation with pseudotumor cerebri also has
been reported. Approximately 50% of patients with the classic form of MSUD develop severe hypoglycemia; this is probably the consequence of a defective gluconeogenesis from amino acids, particularly alanine (118).
been reported. Approximately 50% of patients with the classic form of MSUD develop severe hypoglycemia; this is probably the consequence of a defective gluconeogenesis from amino acids, particularly alanine (118).
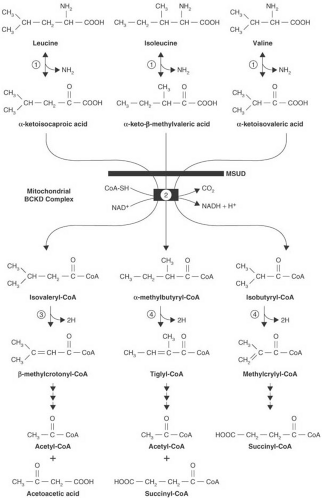 FIGURE 1.5. Degradation of leucine in mammalian tissues. In maple syrup urine disease, the metabolic block is located at step 2. In isovaleric acidemia, the block is confined to step 3. A rare entity with a possible metabolic block at step 4 also has been reported |
MRI during the acute stage of the disease before treatment is characteristic. It demonstrates edema that is maximal in cerebellar deep white matter, the posterior portion of the brainstem, and the posterior limb of the internal capsule. Edema also is seen in the cortical U fibers, the head of the caudate, and the putamen (119,120). These findings are consistent with the location of status spongiosus noted on pathologic examination. The cause of the
acute cerebral edema and its unique localization are unknown. Diffusion MRI suggests that in the myelinated areas of the brain there is an intramyelinic cytotoxic edema, whereas in the unmyelinated areas there is vasogenic interstitial edema (121,122). Because neurologic symptoms become apparent with relatively mild increases in plasma leucine concentrations, whereas there is little apparent toxicity associated with increased levels of isoleucine or valine, damage probably results from leucine accumulation or from accumulation of its ketoacid metabolite. As is the case in PKU, chronic brain damage is probably caused by interference with amino acid transport into the brain, a deranged amino acid environment, and, consequently, failure in biosynthesis of proteolipids, myelin, and neurotransmitters (123). In treated patients, the MRI discloses symmetric bilateral periventricular high signal intensity on T2-weighted images. This picture is similar to that seen in PKU and suggests focal dysmyelination (112).
acute cerebral edema and its unique localization are unknown. Diffusion MRI suggests that in the myelinated areas of the brain there is an intramyelinic cytotoxic edema, whereas in the unmyelinated areas there is vasogenic interstitial edema (121,122). Because neurologic symptoms become apparent with relatively mild increases in plasma leucine concentrations, whereas there is little apparent toxicity associated with increased levels of isoleucine or valine, damage probably results from leucine accumulation or from accumulation of its ketoacid metabolite. As is the case in PKU, chronic brain damage is probably caused by interference with amino acid transport into the brain, a deranged amino acid environment, and, consequently, failure in biosynthesis of proteolipids, myelin, and neurotransmitters (123). In treated patients, the MRI discloses symmetric bilateral periventricular high signal intensity on T2-weighted images. This picture is similar to that seen in PKU and suggests focal dysmyelination (112).
The intermittent form of MSUD results from a variety of mutations in the gene for E2, and branched-chain dehydrogenase activity is higher than in the classic form, usually 5% to 20% of normal (124). The clinical picture is that of intermittent periods of ataxia, drowsiness, behavior disturbances, and seizures that make their first appearance between ages 6 and 9 months. Attacks are generally triggered by infections, immunizations, or other forms of stress (125).
In the intermediate form, the clinical picture is one of mild to moderate mental retardation (126). Branched-chain dehydrogenase activity ranges from 5% to 20% of normal, and the defect is usually in the gene coding for E1 (106).
A thiamin-responsive variant represents an entity in which, in some cases at least, a mutant exists in the gene for E2 (103). Chuang and coworkers proposed that binding of the mutated, inactive E2 component to the wild-type E1 component enhances wild-type E1 activity, and that the augmented E1 activity is responsible for the response to thiamine (114).
Mutants defective in the gene for E3 present with hypotonia, rapid neurologic deterioration, and severe lactic acidosis. This entity is discussed with the various other organic acidemias.
Diagnosis
The most common presentation of MSUD is that of a term infant who initially seems to be well for a few days and then deteriorates. The rate of deterioration varies, and most infants initially are believed to be septic. The neurologist called in to consult on such an infant should always consider the diagnosis of an inborn error of metabolism. Basic investigations at this point include blood or plasma pH, blood gases, glucose, electrolytes, liver function tests, ammonia, and plasma for amino acids and acyl carnitines. Urine for sugars, ketones, and organic acids is also indicated (127). In addition to MSUD, neurologic deterioration during the neonatal period is seen in various organic acidurias, urea cycle defects, fatty acid oxidation defects, and the congenital lactic acidoses. Clinically, MSUD is distinguished by the characteristic odor of the patient, a positive urine 2,4-dinitrophenylhydrazine test, and an elevation of the plasma branched-chain amino acids. The characteristic increase in plasma leucine, isoleucine, and valine is seen by the time the infant is 24 hours old, even in those infants who have not yet been given protein (128). Routine newborn screening for the condition using a bacterial inhibition assay analogous to that used for the neonatal diagnosis of PKU is performed in many states in the United States and in many other countries (129). Tandem mass spectroscopy also can be used and has the advantage of obtaining rapid quantitative measurements of all three branched-chain amino acids (43). The presence of the branched-chain ketoacid decarboxylases in cultivated amniocytes and chorionic villi allows the antenatal diagnosis of the disease as early as 10 weeks’ gestation (130).
Treatment
Treatment consists in inhibiting endogenous protein catabolism, sustaining protein synthesis, preventing deficiencies of essential amino acids, and maintaining normal serum osmolarity (131). Morton and coworkers stressed that restriction of the dietary intake of the branched-chain amino acids through the use of one of several commercially available formulas is secondary in importance to inhibition of protein catabolism and enhancement of protein synthesis (131). For optimal results, infants should be placed on the diet during the first few days of life and should receive frequent measurements of serum amino acids. Prompt and vigorous treatment of even mild infections is mandatory; a number of children on this synthetic diet have died of septicemia (115,131).
Peritoneal dialysis or hemodialysis has been used to correct coma or other acute neurologic symptoms in the newly diagnosed infant (132). Another, simpler approach is to provide intravenously or by nasogastric tube an amino acid mixture devoid of leucine but containing large amounts of tyrosine, glutamine, and alanine (133). Morton and coworkers believe that the brain edema that is frequently seen in the acutely ill neonate results from hyponatremia and responds promptly to addition of oral and intravenous saline (131). Most of the children in whom long-term dietary therapy was initiated during the first 2 weeks of life and whose dietary control was meticulously maintained achieved normal or nearly normal IQs (131,134). In the experience of Hilliges and coworkers, the mean IQ of MSUD patients at 3 to 16 years of age was 74 ± 14, as compared with 101 ± 12 for early-treated patients with PKU. The length of time after birth that plasma leucine
concentrations remain elevated appears to affect the IQ, as does the amount, if any, of residual branched-chain ketoacid dehydrogenase activity (135). The thiamin-responsive child is treated with 10 to 1,000 mg of thiamin per day (103).
concentrations remain elevated appears to affect the IQ, as does the amount, if any, of residual branched-chain ketoacid dehydrogenase activity (135). The thiamin-responsive child is treated with 10 to 1,000 mg of thiamin per day (103).
Nonketotic Hyperglycinemia (OMIM 238300)
This relatively common family of diseases is marked by genetic and phenotypic heterogeneity, and considerable variation occurs in the severity of neurologic symptoms (136).
Five forms have been recognized:
In the most common, infantile form, neurologic symptoms begin during the neonatal period. They are highlighted by profound hypotonia, intractable generalized, reflex, or myoclonic seizures, apnea, and progressive obtundation with coma and respiratory arrest. The EEG demonstrates a burst-suppression pattern or, later, hypsarrhythmia (137). Nystagmus and a marked abnormality of the electroretinogram (ERG) also are seen (138). The majority of affected infants die during the neonatal period; those who survive are profoundly retarded. In some infants acute hydrocephalus develops between 2 and 6 months. In all instances this has been associated with a large retrocerebellar cyst (139).
A transient neonatal form has been recognized that initially is clinically indistinguishable from the permanent form of nonketotic hyperglycinemia. However, symptoms remit abruptly after a few days or months, and youngsters are left unimpaired (140). The condition appears to develop in some heterozygous carriers for nonketotic hyperglycinemia (141), and subsequent development is normal (142).
A less severe form becomes apparent during the latter part of the first year of life after several months of normal development. It is marked by progressive dementia leading to decerebrate rigidity. Extrapyramidal signs are not uncommon (143).
A juvenile form with mild mental retardation, hyperactivity, and language deficits also has been reported. They may represent survival to adulthood of individuals with the mild infantile form (144).
Bank and Morrow reported adults with a clinical picture of weakness and spasticity resembling spinocerebellar ataxia (145).
Pathologic examination of the brain in the infantile form of the disease discloses a reduction in white matter with an extensive spongy degeneration accompanied by marked gliosis (146). Partial or complete agenesis of the corpus callosum has been described, an indication of a significant intrauterine insult (137).
The marked increase in plasma and CSF glycine and the markedly elevated ratio of CSF glycine to blood glycine are diagnostic of the condition (137). It is important to note that one cannot rely on the plasma glycine alone to arrive at a diagnosis. The MRI shows decreased or absent supratentorial white matter, with thinning of the corpus callosum and cortical atrophy (147). On diffusion-weighted MRI symmetric lesions in the dorsal brainstem, cerebral peduncles, and posterior limbs of the internal capsule are noted, a picture compatible with a vacuolating myelopathy (148).
The basic defect in this condition is localized to the mitochondrial glycine cleavage system, which converts glycine to serine and is expressed in liver and brain. This complex reaction requires four protein components, and defects in one or another of three of these components have been documented (137). Some correlation exists between the clinical expression of the disease and the genetic defect. The classic neonatal form of the disease usually is associated with virtual absence of the pyridoxal-containing decarboxylase (P protein) and the milder atypical forms with a defect in the tetrahydrofolate-requiring transfer protein (T protein) (137,149).
The pathophysiology of the neurologic abnormalities has not been established fully. Glycine is an inhibitory neurotransmitter that acts mainly at spinal cord and brainstem levels. It also acts as a coagonist for the N-methyl-D-aspartate glutamate receptor, modulating its activity and probably producing seizures by an excitotoxic mechanism (137). The inhibitory effects of glycine are blocked by strychnine, but the effectiveness of strychnine on the basic course of the illness is questionable. Blockers of the N-methyl-D-aspartate receptor, such as dextromorphan or ketamine, have been used in conjunction with sodium benzoate, which is intended to couple with glycine. Despite these interventions the outcome for the neonatal form of nonketotic hyperglycinemia is dismal (150,151). If the patients survive the first 2 weeks of intubation, the apnea often resolves.
Defects in Urea Cycle Metabolism
Six inborn errors in the urea cycle have been described. Five of these represent a lesion at each of the five steps in the conversion of ammonia to urea (Fig. 1.6). These include argininosuccinic aciduria, citrullinuria, hyperargininemia, and two conditions termed hyperammonemia, the more common one attributable to a defect of ornithine transcarbamylase (OTC) and the other the result of a defect in mitochondrial carbamyl phosphate synthetase (CPS). The genes for all components of the urea cycle have been cloned. Additionally, a deficiency of N-acetylglutamate synthetase has been reported (152). This enzyme is responsible for the formation of N-acetylglutamate, a required activator for mitochondrial CPS. More recently two genetic defects affecting the citrulline and ornithine transporters have also been documented. The various deficits are summarized in Table 1.10.
The biochemical aspects of the urea cycle were reviewed by Batshaw (153).
The biochemical aspects of the urea cycle were reviewed by Batshaw (153).
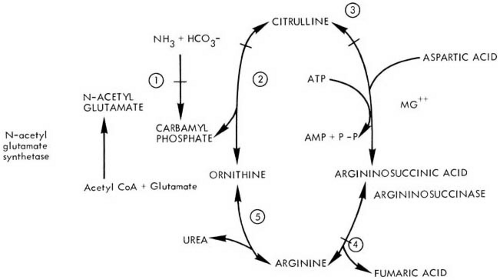 FIGURE 1.6. Normal urea cycle. In argininosuccinic aciduria, the cycle is blocked at step 4. In citrullinuria, the block occurs at step 3. In ornithine transcarbamylase deficiency, the block is at step 2. In carbamoylphosphate synthetase deficiency, the block is at step 1. A defect in N-acetylglutamate synthetase results in hyperammonemia by depriving step 1 of its activator, N-acetylglutamate. The enzyme is inhibited by a variety of organic thioesters, notably propionyl-CoA and isovaleryl-CoA. In hyperargininemia, the defect is one of arginase, step 5. |
Because most systemic and neurologic symptoms in these diseases are the consequences of hyperammonemia or the accumulation of urea cycle intermediates, clinical manifestations of the urea cycle defects are nonspecific and overlap considerably. In their classic presentation, which occurs in some 60% of cases (154), the conditions become apparent between 24 and 72 hours of life. Initial symptoms include vomiting, lethargy, hyperpnea, and
hypotonia. These progress rapidly to seizures and coma. The EEG often shows burst suppression, and neuroimaging indicates the presence of cerebral edema.
hypotonia. These progress rapidly to seizures and coma. The EEG often shows burst suppression, and neuroimaging indicates the presence of cerebral edema.
TABLE 1.10 Disorders of Urea Cycle and of Ornithine | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
When the enzyme deficiency is less severe, hyperammonemic episodes are delayed to late infancy or childhood. Patients have recurrent episodes of lethargy, vomiting, and, less often, seizures. Hyperactivity, behavioral abnormalities, and moderate to severe mental retardation are common, as is intolerance of protein-containing foods (155).
Argininosuccinic Aciduria (OMIM 207900)
Argininosuccinic aciduria is one of the more common of the urea cycle disorders. The condition is characterized by mental retardation, poorly formed hair, and accumulation of argininosuccinic acid in body fluids. It was first described in 1958 by Allan and coworkers (156).
Molecular Genetics and Biochemical Pathology
Argininosuccinic acid is a normal intermediary metabolite in the synthesis of urea (see Fig. 1.6). A deficiency in argininosuccinate lyase, an enzyme whose gene has been mapped to chromosome 7cn–q11.2, has been demonstrated in liver and skin fibroblast cultures (157).
The synthesis of urea is only slightly depressed, but a large proportion of labeled ammonium lactate administered to affected individuals is converted to glutamine (158). The manner in which children synthesize urea is not clear. It appears likely that in argininosuccinic aciduria, as well as in the other defects of urea cycle, substrate accumulates to a concentration at which the decreased substrate-binding capacity of the mutant enzyme is overcome by the accumulation of precursor to levels greater than the KM for the mutated enzyme (159).
Pathologic Anatomy
The liver architecture is abnormal, with increased fat deposition. The brain of a neonate who succumbed to the disease was edematous, with poor demarcation of gray and white matter. The cortical layers were poorly developed, and myelination was defective with vacuolated myelin sheaths and cystic degeneration of white matter (160). An older patient had atypical astrocytes similar to the Alzheimer II cells seen in Wilson disease and in severe chronic liver disease (161).
Clinical Manifestations
As ascertained by newborn screening, the incidence of argininosuccinic aciduria is 1 in 70,000 in Massachusetts and 1 in 91,000 in Austria (162). Three distinct clinical forms have been recognized, each resulting from a different genetic mutation (163).
The most severe entity is the neonatal form. Infants feed poorly, become lethargic, develop seizures, and generally die within 2 weeks (160,164). In a second form, progression is less rapid, but similar symptoms appear in early infancy. In the majority of patients, including those described by Allan and coworkers (156), the presenting symptoms are mental retardation, recurrent generalized convulsions, poorly pigmented, brittle hair (trichorrhexis nodosa), ataxia, and hepatomegaly (165). Some patients have been seizure free, however, and have presented with little more than learning difficulties, and others (approximately 20% of all affected children) have normal intelligence without treatment (166).
Diagnosis
The presence of elevated blood ammonia should suggest a disorder in the urea cycle. Initial evaluation of such a child should include routine blood chemistries, plasma lactate levels, liver function tests, quantitative assay of plasma amino acids, and assay of urine for organic acids and orotic acid (153). The specific diagnosis of argininosuccinic aciduria can be made by a significant elevation of plasma citrulline and the presence of large quantities of plasma, urinary, and CSF argininosuccinic acid. In some instances, fasting blood ammonia level can be normal or only slightly elevated, but marked elevations occur after protein loading. Increased excretion of orotic acid is seen in all urea cycle defects, with the exception of CPS deficiency (167).
Treatment
Hyperammonemic coma in the neonate caused by any of the urea cycle defects requires prompt intervention. In essence, treatment consists of detoxification and removal of the excess ammonia and reduction in the formation of ammonia. Quantitative amino acid chromatography should be performed on an emergency basis, and the infant should be given a high dose of intravenous glucose with insulin to suppress protein catabolism. The elevated ammonia levels can be reduced by hemodialysis if available or by peritoneal dialysis (168). Details of treatment are presented by Brusilow and Horwich (166) and Batshaw and colleagues (169). Treatment for increased intracranial pressure, which frequently accompanies neonatal hyperammonemia, is symptomatic.
The long-term management of an infant who suffers a urea cycle defect is directed toward lowering blood ammonia levels and maintaining them as close to normal as possible. This is accomplished by providing the infant with alternative pathways for waste nitrogen synthesis and excretion. Infants with argininosuccinic aciduria are placed on a protein-restricted diet (1.2 to 2.0 g/kg per
day), which is supplemented with L-arginine (0.4 to 0.7 g/kg per day), which promotes the synthesis of citrulline and argininosuccinate as waste nitrogen products, and citrate, which improves weight gain and reduces hepatomegaly (166,169). Sodium phenylbutyrate is added to divert ammonia from the urea cycle. A number of centers are now recommending the use of liver transplantation for argininosuccinic aciduria (170).
day), which is supplemented with L-arginine (0.4 to 0.7 g/kg per day), which promotes the synthesis of citrulline and argininosuccinate as waste nitrogen products, and citrate, which improves weight gain and reduces hepatomegaly (166,169). Sodium phenylbutyrate is added to divert ammonia from the urea cycle. A number of centers are now recommending the use of liver transplantation for argininosuccinic aciduria (170).
Episodes of hyperammonemia are generally triggered by intercurrent infections. Prevention of a rapid progression to death requires hospitalization and the use of intravenous therapy (166).
On this regimen, some children with argininosuccinic aciduria do well. Reduction of blood ammonia levels is accompanied by improved growth, reduction in liver size, cessation of seizures, and, in some patients, normal hair. Intellectual function is significantly impaired, however. In the experience of Batshaw and coworkers, all infants with the severe, neonatal form of the disease survived, although those who have been followed the longest have shown a significant lowering of their IQ (169). In the French long-term follow-up of 15 patients with argininosuccinic aciduria, none were doing well (171). Children with the late-onset variant fare much better, and, with therapy, achieve normal development (165). As a rule, the individual’s ultimate IQ is a function of the severity and duration of hyperammonemic coma, and children who have not recovered from coma within 5 days do poorly (172). Valproic acid cannot be used in the treatment of seizures associated with this and with the other urea cycle defects because it induces severe hyperammonemia at even low doses (173).
Citrullinemia
In 1963, McMurray and associates reported a mentally retarded infant who had a metabolic block in the conversion of citrulline to argininosuccinic acid (see Fig. 1.6, step 3) (174). Since then, it has become clear that this condition, like many of the other inborn metabolic errors, is heterogeneous. Two genotypically and phenotypically distinct conditions have been recognized.
Argininosuccinic Acid Synthetase Deficiency (CTLI) (OMIM 215700)
The gene coding for argininosuccinic acid synthetase has been cloned. It is carried on chromosome 9 (175). At least 50 different genetic mutations have been recorded for infants with neonatal citrullinemia (176).
As a result of the enzymatic defect, the concentration of citrulline in urine, serum, and CSF is markedly increased, and administration of a protein meal results in a dramatic increase of blood ammonia and urinary orotic acid. Blood and urinary urea values are normal, indicating that urea production is not completely blocked. CT and MRI studies performed on patients with the neonatal form of citrullinemia show lesions in the thalami, basal ganglia, cortex, and subcortical white matter. Diffusion-weighted MR images indicate the presence of cytotoxic edema. Follow-up studies reveal subcortical cysts, ulegyria, and atrophy (177,178).
In Western countries, the most common presentation is in the neonatal period with lethargy, hypotonia, and seizures (179). In other instances, the disease is less severe, even though recurrent bouts of vomiting, ataxia, and seizures can start in infancy. A third form presents with mental retardation. Completely asymptomatic individuals also have been encountered (174,179).
Treatment for citrullinemia is similar to treatment for argininosuccinic aciduria, except that for long-term therapy, the low-protein diet is supplemented with arginine and sodium phenylbutyrate (166,169).
Citrullinuria in the absence of citrullinemia has been seen in patients with cystinuria. In this instance, citrulline is derived from arginine, which is poorly absorbed from the intestine (180).
Adult-Onset Type II Citrullinemia
Late-onset citrullinemia with loss of enzymatic activity in liver, but not in kidney or fibroblasts, is seen predominantly in Japan, where it constitutes the most common form of citrullinemia (181). It presents with cyclical changes in behavior, dysarthria, and motor weakness. It is due to mutations in citrin, a mitochondrial aspartate glutamate carrier (182,183).
Ornithine Transcarbamylase (OTC) Deficiency (OMIM 311250)
OTC is an enzyme coded by an X-linked gene and located in the mitochondrial matrix. Deficiency of OTC in the male infant is characterized biochemically by a catastrophic elevation of blood ammonia. This is accompanied by an increased excretion of orotic acid and a generalized elevation of plasma and urine amino acids. The disease was first reported in 1962 by Russell and coworkers (184) and is the most common of the urea cycle defects. The gene coding for the enzyme has been cloned and localized to the short arm of the X chromosome (Xp21.1), close to the Duchenne muscular dystrophy locus, and most families have their own unique mutation (185). The enzyme defect can be complete or, as occurs in some 10% to 20% of hemizygous male patients, it can be partial (186). As a consequence, blood ammonia levels are strikingly and consistently elevated (0.4 to 1.0 mg/dl, or 230 to 580 μmol/L, contrasted with normal values of less than 0.1 mg/dL, or 50 μmol/L), and CSF ammonia is at least 10 times normal. Additionally, there is an accumulation of glutamine, glutamate, and alanine. This is accompanied by a striking reduction in plasma citrulline and an increased excretion
of orotic acid. The last is the consequence of a diffusion of excess carbamyl phosphate from mitochondria into cytosol, where it is converted into orotic acid (187).
of orotic acid. The last is the consequence of a diffusion of excess carbamyl phosphate from mitochondria into cytosol, where it is converted into orotic acid (187).
As is the case in argininosuccinic aciduria, the neuropathologic picture is highlighted by the presence of Alzheimer II astrocytes throughout the brain (188). Unlike hepatic encephalopathy, a striking degree of neuronal necrosis also exists. Electron microscopic examination of liver can reveal striking abnormalities of the mitochondria (189).
As a rule, the magnitude of the enzymatic defect correlates with the severity of clinical symptoms. In male patients, the clinical picture is marked by severe hyperammonemia. When the condition presents during the neonatal period it is rapidly progressive, with a high incidence of mortality or profound neurologic residua. Symptoms usually are delayed until the second day of life and are highlighted by feeding difficulties, lethargy, and respiratory distress. The plasma ammonia level is at least five times normal, thus distinguishing the condition from sepsis (190). MRI demonstrates injury to the lentiform nuclei and the deep sulci of the insular and perirolandic region (191).
Less severe cases present with failure to thrive and with episodic attacks of headache and vomiting followed by periods of lethargy and stupor. These attacks are often the consequence of protein ingestion and are accompanied by high blood ammonia levels (192). Although hyperammonemia is probably responsible for a considerable proportion of the neurologic symptoms, alterations in neurotransmitters, notably quinolinic acid, a known excitotoxin that accumulates as a result of increased tryptophan transport across the blood–brain barrier, also could be involved (193).
The disease is expressed more variably in the heterozygous female patient, with manifestations ranging from apparent normalcy to profound neurologic deficits (187). In symptomatic female patients, behavioral abnormalities are almost invariable. In the series of Rowe and coworkers, irritability, temper tantrums, inconsolable crying, and hyperactivity were seen in every patient (187). Episodic vomiting and lethargy were also invariable. Ataxia was seen in 77% of female patients, reduced physical growth in 38%, and developmental delay in 35%. Seizures, generalized or focal, were seen in 23% (187). Blood ammonia and urinary orotic acid levels were elevated consistently when girls were symptomatic. Other girls are asymptomatic except for an aversion to protein-rich foods and possible subtle cognitive deficits (193). In some women the first hyperammonemic episode may occur in the postpartum period (194). Valproate therapy can induce fatal hepatotoxicity in male patients with OTC deficiency and in heterozygous female patients (195).
Treatment of OTC deficiency in the male or female patient is similar to treatment for argininosuccinic aciduria. It is directed at decreasing protein intake by means of a low-protein diet and increasing waste nitrogen excretion by the addition of sodium phenylbutyrate and arginine or citrulline to the diet (196). Liver transplant or isolated hepatocyte transplant has been suggested for the severe neonatal form, but the outcome for infants with no significant OTC activity is poor (171,197). Prospective treatment of infants at risk for neonatal OTC deficiency has been attempted with some success in that such infants appear to have a better neurologic outcome than those who have to be rescued from hyperammonemic coma (198).
In some hemizygous male patients, OTC deficiency is not complete, and the clinical course is not as severe. It can consist of several months of normal development followed by progressive cerebral degeneration or by the acute onset of cerebral and hepatic symptoms resembling those of Reye syndrome (199).
OTC is expressed only in liver and in the small intestine; prenatal diagnosis therefore depends on mutation detection or linkage analysis. Because some cases represent new mutations, linkage analysis is of limited use, except for offspring of obligate gene carriers (200). Of course one cannot predict whether a female patient will be asymptomatic or severely affected (186).
Carbamyl Phosphate Synthetase (CPS) Deficiency (OMIM 237300)
Carbamyl phosphate synthetase deficiency is a disorder of the urea cycle manifested by a reduction in hepatic mitochondrial CPS activity (see Fig. 1.6, step 1). This condition was reported first by Freeman and coworkers (201).
Symptoms of CPS deficiency are the most severe of any of the urea cycle defects, and the neonatal form of the condition, which is associated with complete absence of the enzyme, is usually fatal. In partial CPS deficiency, symptoms appear in infancy and consist of recurrent episodes of vomiting and lethargy, convulsions, hypotonia or hypertonia, and irregular eye movements (202). Imaging studies show changes that are almost identical to those seen in OTC deficiency. During the acute state there is cerebral edema (191,202).
Autopsy reveals ulegyria of cerebral and cerebellar cortex and hypomyelination of the centrum semiovale and the central part of the brainstem. In contrast to argininosuccinic aciduria, no Alzheimer cells are seen, probably because these cells take some time to develop and CPS deficiency is usually rapidly fatal (186,203).
Carbamyl phosphate synthetase deficiency is diagnosed by the presence of hyperammonemia in the absence of an elevation of plasma citrulline, argininosuccinic acids, or arginine. In contrast to OTC deficiency, orotic acid excretion is low or normal. Treatment for the neonatal and the less severe older-onset forms of CPS deficiency is similar to that for OTC deficiency, but the outcome in the neonatal form is uniformly poor (166,172).
Hyperargininemia (OMIM 207800)
Hyperargininemia, the least common of urea cycle disorders, has a clinical picture that differs from that of the other urea cycle disorders in that there is infrequent hyperammonemia. In the acute form cerebral edema and seizures begin during the neonatal period (204). In other instances, mental retardation, microcephaly, spastic diplegia, or quadriparesis becomes apparent during the first few months or years (205). The concentration of glutamine and arginine in plasma and CSF is elevated, and excretion of arginine, cystine, and lysine is increased, a urinary amino acid pattern resembling that of cystinuria. Blood ammonia levels are normal or slightly elevated. This is probably due to the presence of a second unaffected arginase gene locus expressed primarily in kidney mitochondria. A deficiency of arginase has been documented in red cells and liver (206).
Patients are treated with a diet consisting of a mixture of essential amino acids, exclusive of arginine, and supplemented by a commercial formula that furnishes fats, carbohydrates, vitamins, and minerals. Phenylbutyrate can also be added to the regimen. Replacement of arginase by means of periodic exchange transfusions has been suggested as a supplementary means of controlling blood and CSF arginine concentrations (207). In spite of therapy, spasticity often progresses. The effectiveness of liver transplants is unknown.
N-Acetylglutamate Synthetase Deficiency (OMIM 237310)
In the absence of the enzyme, a deficiency of N-acetylglutamate exists, an activator of mitochondrial CPS (185). Clinical manifestations range from fatal neonatal hyperammonemia to protein intolerance with recurrent episodes of hyperammonemia (208,209a). Treatment with carbamylglutamate has been successful.
Other Genetic Causes of Hyperammonemia
Hyperammonemia is seen in several other genetic disorders. Hyperammonemia, with increased excretion of orotic acid, is seen in periodic hyperlysinemia. For reasons as yet unknown, hyperammonemia can be induced by administration of large amounts of lysine and the condition diagnosed by the excretion of large amounts of lysine. It is considered with the other defects of lysine metabolism.
Ornithinemias
Another cause for intermittent hyperammonemia is ornithinemia. Clinically and biochemically this is a heterogeneous entity, At least two conditions have been delineated.
HHH Syndrome (OMIM 238970)
This condition is caused by a mutation in a gene encoding a mitochondrial ornithine transporter (210). As the name indicates, elevated plasma ornithine levels (hyperornithinemia) are accompanied by hyperammonemia and homocitrullinuria. In the neonatal form the clinical picture is of prolonged neonatal jaundice, mental retardation, infantile spasms, and intermittent ataxia (211,212). When the condition becomes apparent later in life, it is marked by spastic gait, myoclonic seizures, and ataxia (200).
Ornithine Aminotransferase Deficiency (OMIM 258870)
In this entity, ornithinemia is accompanied by gyrate atrophy of the choroid and retina, leading to night blindness. There is no hyperammonemia. The condition is most commonly encountered in the Finnish population, in which there is an incidence of 1 in 50,000. Intelligence is preserved, and no obvious neurologic or muscular symptoms occur, although type 2 muscle fiber atrophy is seen on biopsy (213) and peripheral nerve involvement can be shown electrically (214). Early white matter degenerative changes and premature cerebral atrophy can be documented on MRI, and MRS demonstrates reduced creatine in muscle and brain (215,216). The creatine deficiency can be partially corrected by creatine supplementation (216). Ocular symptoms seem to be ameliorated by a low-arginine diet or creatine supplementation.
Other Causes for Hyperammonemia
Transient hyperammonemia with consequent profound neurologic depression can be encountered in asphyxiated infants or with significant dehydration (217,218). This state should be differentiated not only from the various urea cycle defects, but also from the various organic acidemias, notably methylmalonic acidemia and propionic acidemia, which can induce hyperammonemia (219). In these conditions, the accumulation of organic acids inhibits the formation of N-acetylglutamine, the activator of mitochondrial CPS, and the activities of all five enzymes of the urea cycle are depressed. On a clinical basis, the organic acidemias can be distinguished from urea cycle defects in that infants with a urea cycle defect are asymptomatic for the first 24 hours of life and only rarely develop coma before 72 hours. Additionally, they demonstrate tachypnea rather than a respiratory distress syndrome. In contrast to the distressed neonates with hyperammonemia, infants with the various organic acidemias demonstrate ketonuria or ketonemia as well as acidosis (220).
Asymptomatic hyperammonemia is relatively common in low-birth-weight neonates. It probably is caused by shunting of blood away from the portal circulation of the liver into the systemic circulation.
 FIGURE 1.7. Normal metabolism of sulfur amino acids. THF, tetrahydrofolate. The known genetic defects that cause homocystinuria are a deficiency of cystathionine-β-synthase (1), N5,10-methylenetetrahydrofolate reductase (2), methionine synthase or methionine synthase reductase (3), or deficient synthesis of methylcobalamine (4). Other defects in the pathway result in cystathioninuria due to γ-cystathionase deficiency (7), sulfite oxidase and molybdenum cofactor synthesis deficiencies (8), and hyperammonemia due to methionine adenosyltransferase deficiency (6). Betaine can be given therapeutically to treat homocystinuria by increasing remethylation of homocysteine via betaine-homocysteine methyltransferase (5). (From Wilcox WR, Cedarbaum S. Amino acid metabolism. In: Rimoin DL, Connor JM, Pyeritz RE, et al., eds. Principles and practice of medical genetics, 4th ed. New York: Churchill Livingstone, 2002;2405–2440.) |
Mild hyperammonemia can also be seen in associated with hypoglycemia and hyperinsulinism due to mutations in the glutamate dehydrogenase gene (221).
Defects in the Metabolism of Sulfur Amino Acids
Homocystinuria (OMIM 236200)
The increased excretion of homocystine is a manifestation of several inborn errors of methionine metabolism. The most common of these errors is marked by multiple thromboembolic episodes, ectopia lentis, and mental retardation. Although discovered as late as 1962 by Field and reported subsequently by Carson and coworkers (222), the prevalence of homocystinuria varies considerably from one country to another, ranging from 1 in 65,000 in Ireland to approximately 1 in 335,000 worldwide (223). The gene for this autosomal recessive condition has been cloned; it is localized to the long arm of chromosome 21 (21q22.3).
Molecular Genetics and Biochemical Pathology
In the most common genetic form of homocystinuria, the mutation involves the gene for cystathionine synthase, the enzyme that catalyzes the formation of cystathionine from homocysteine and serine (Fig. 1.7) (224). The enzyme as purified from human liver has two identical subunits and contains bound pyridoxal phosphate (225). Considerable genetic heterogeneity exists among various cystathionine synthase–deficient families, but in the majority, the lesion resides in a structural gene for the enzyme (226). In most homocystinuric patients, the mutation does not cause dysfunction of the catalytic domain of this enzyme, but instead interferes with its activation by pyridoxine (227). As a result, enzyme activity is either
completely absent or, as is the case in a significant proportion of affected families (in the Australian series of Gaustadnes and coworkers approximately 38% of patients with homocystinuria), residual activity occurs (228). In the latter group, addition of pyridoxine (500 mg/day or more) stimulates enzyme activity and partially or completely abolishes the excretion of homocystine, the oxidized derivative of homocysteine. Pyridoxine-responsive patients tend to have a milder phenotype of the disease.
completely absent or, as is the case in a significant proportion of affected families (in the Australian series of Gaustadnes and coworkers approximately 38% of patients with homocystinuria), residual activity occurs (228). In the latter group, addition of pyridoxine (500 mg/day or more) stimulates enzyme activity and partially or completely abolishes the excretion of homocystine, the oxidized derivative of homocysteine. Pyridoxine-responsive patients tend to have a milder phenotype of the disease.
TABLE 1.11 Disorders of Sulfur Amino Acids | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
As a result of the block, increased amounts of homocystine, the oxidized derivative of homocysteine, and its precursor, methionine, are found in urine, plasma, and CSF. Administration of a methionine load to affected individuals produces a striking and prolonged increase in plasma methionine, but little alteration in the homocystine levels. In part, this reflects the low renal threshold for homocystine.
The various other genetic defects that result in an increased excretion of homocystinuria are depicted in Table 1.11. Homocystinuria also can result from an impaired methylation of homocysteine to methionine (see Fig. 1.7). When the metabolic block is at this point, plasma methionine concentrations are normal rather than increased, as is the case in the more common form of homocystinuria owing to cystathionine synthase deficiency. Methylation uses N5-methyltetrahydrofolate as a methyl donor and a vitamin B12 derivative (methylcobalamin) as a cofactor. Methylation can be impaired as a result of lack
of the cofactor methylcobalamin, or the enzyme. When synthesis of the vitamin B12 cofactor is defective, the biochemical picture is characterized by increased excretion of methylmalonic acid and homocystine. This condition is distinct from methylmalonic aciduria (MMA), which is the result of a reduced activity of methylmalonyl-CoA mutase, a cobalamin-dependent enzyme. Several errors in cobalamin metabolism have been recognized; the clinical picture includes mental retardation, seizures, failure to thrive, hypotonia, ataxia, and megaloblastic anemia. The conditions are covered in the section on Organic Acidurias, later in this chapter.
of the cofactor methylcobalamin, or the enzyme. When synthesis of the vitamin B12 cofactor is defective, the biochemical picture is characterized by increased excretion of methylmalonic acid and homocystine. This condition is distinct from methylmalonic aciduria (MMA), which is the result of a reduced activity of methylmalonyl-CoA mutase, a cobalamin-dependent enzyme. Several errors in cobalamin metabolism have been recognized; the clinical picture includes mental retardation, seizures, failure to thrive, hypotonia, ataxia, and megaloblastic anemia. The conditions are covered in the section on Organic Acidurias, later in this chapter.
Another cause for homocystinuria is a defect in the methylation enzyme, methylene tetrahydrofolate reductase (MHTFR). The clinical picture in these patients is protean. Some are retarded or have been diagnosed as schizophrenic; others have recurrent episodes of vomiting and lethargy or muscular weakness and seizures. Vascular thromboses also have been encountered, but the skeletal and ocular changes of homocystinuria are absent (229,230). Folic acid has reduced the biochemical abnormalities in some patients with this condition but has been ineffectual in others. This severe disorder due to marked deficiency in MHTFR should be distinguished from the common “thermolabile” variant that mildly increases plasma homocysteine levels.
Pathologic Anatomy
The primary structural alterations in homocystinuria are noted in blood vessels of all calibers (231). Most of these show intimal thickening and fibrosis; in the aorta and its major branches, fraying of elastic fibers might be observed. Arterial and venous thromboses are common in a number of organs. Within the brain are usually multiple infarcted areas of varying age. The existence of dural sinus thrombosis has been recorded.
How the metabolic defect induces a propensity to vascular thrombosis has been reviewed by Welch and Loscalzo (232). It has also become evident that an increased plasma homocysteine concentration is an independent risk factor for atherosclerotic vascular disease.
Clinical Manifestations
The pyridoxine-unresponsive form of homocystinuria is more severe in its manifestations than the pyridoxine-responsive form.
Homocystinuric infants appear healthy at birth, and their early development is unremarkable until seizures, developmental slowing, or cerebrovascular accidents occur between 5 and 9 months of age. Ectopia lentis is seen in more than 90% of affected individuals. Lenticular dislocation has been recognized as early as age 18 months, but it generally occurs between 3 and 10 years of age. The typical older homocystinuric child’s hair is sparse, blond, and brittle, and multiple erythematous blotches are seen over the skin, particularly across the maxillary areas and cheeks. The gait is shuffling, the extremities and digits are long, and genu valgum is present in most instances. Secondary glaucoma and cataracts are common (233).
In approximately 50% of the patients reported, major thromboembolic episodes have occurred on one or more occasions. These include fatal thromboses of the pulmonary artery, coronary arteries, and renal artery and vein. Multiple major cerebrovascular accidents also result in hemiplegia, and ultimately in a picture that closely resembles pseudobulbar palsy. Thromboembolic events are particularly common after even minor surgical procedures. It is likely that minor and unrecognized cerebral thrombi are the cause of the mental retardation that occurs in 50% of the patients (234,235). Routine laboratory study results are normal, but in a high proportion of patients, electromyography suggests myopathy (235).
Radiography reveals a biconcavity of the posterior aspects of the vertebrae (codfish vertebrae) (236). Additionally, scoliosis and osteoporosis become apparent in late childhood. Abnormalities of the hands and feet are noted also. These include metaphyseal spicules, enlargement of carpal bones, and selective retardation of the development of the lunate bone (237). Neuroimaging studies tend to show lesions due to vascular ischemia.
Diagnosis
The diagnosis of homocystinuria suggested by the appearance of the patient can be confirmed by the increased urinary excretion of homocystine, by elevated plasma methionine and homocystine, and by a positive urinary cyanide-nitroprusside reaction. Enzyme activity can be determined in cultured skin fibroblasts or in liver biopsy specimens.
Although ectopia lentis, arachnodactyly, and cardiovascular symptoms are seen also in Marfan syndrome, homocystinuria can be distinguished by its autosomal recessive transmission (in contrast to the dominant transmission of Marfan syndrome), the thromboembolic phenomena, the early appearance of osteoporosis, the biconcave vertebrae, and the peculiar facial appearance (238). The relatively long fingers seen in Marfan syndrome are present at birth, and the skeletal disproportion remains constant. In homocystinuria, the skeleton is normal for the first few years of life, but the limbs grow disproportionately long. Ectopia lentis is seen not only in homocystinuria but also as an isolated congenital defect in the Weill-Marchesani syndrome and in sulfite oxidase deficiency. In the latter condition, it occurs in conjunction with profound mental retardation, seizures commencing shortly after birth, acute hemiplegia, opisthotonos, and hyperacusis. Because the majority of cases are the result of a deficiency of the molybdenum cofactor rather than of the apoenzyme, this condition is covered more fully in the section dealing with disorders of metal metabolism.
Cystathionine synthase has been found in cultivated amniotic fluid cells, and the condition, therefore, can be diagnosed prenatally (239).
Treatment
Restriction of methionine intake lowers plasma methionine and eliminates the abnormally high urinary excretion of homocystine. Commercially available diets that are methionine free and are supplemented by carbohydrates, fats, and fat-soluble vitamins generally lower plasma methionine levels to the normal range (235). The diets are supplemented with cystine.
Other dietary measures include the addition of folic acid, based on the assumption that the mental defect is in part related to low serum folate levels (240). Dietary supplementation with betaine hydrochloride (N,N,N-trimethylglycine), a methyl donor, is generally used in pyridoxine-nonresponsive patients (241). However, several instances of progressive cerebral edema have been encountered in patients whose serum methionine levels had not been well controlled (242). Antithrombotic agents, such as aspirin or dipyridamole, are also given, although their effectiveness has not been proven.
Early therapy with good biochemical control results in a normal IQ and significantly reduces the incidence of thromboembolic episodes and other vascular complications in pyridoxine-nonresponsive patients (243,244).
In pyridoxine-responsive patients, large doses of the vitamin (250 to 1,200 mg/day) reduce or eliminate biochemical abnormalities. In the series of Mudd and coworkers, pyridoxine-responsive patients treated from the neonatal period on had an IQ ranging from 82 to 110. Virtually all patients with IQs greater than 90 were found to be responsive to pyridoxine (245).
Hypermethioninemia
Several other conditions are marked by elevated plasma methionine levels. The most common of these is a transitory methioninemia, seen in infants, many of who are premature and receiving a high-protein diet (at least 7 g/kg per day). It is likely that the biochemical abnormality is caused by delayed maturation of one or more of the enzymes of methionine metabolism.
Methioninemia, with or without tyrosinemia, accompanied by hepatorenal disease (tyrosinemia I) is considered in the section on tyrosinosis and tyrosinemia (see Table 1.9).
Persistent methioninemia associated with a deficiency of hepatic methionine adenosyltransferase is a benign metabolic variant unaccompanied by neurologic symptoms or impairment of cognition (246).
Other Rare Metabolic Defects
A few other extremely rare defects of amino acid metabolism associated with neurologic symptoms are presented in Table 1.12. Experience with disorders such as the iminoacidemias, cystathioninuria, and histidinemia should caution the reader against accepting a causal relationship between metabolic and neurologic defects.
DISORDERS OF RENAL AMINO ACID TRANSPORT
Renal amino acid transport is handled by five specific systems that have nonoverlapping substrate preferences. The disorders that result from genetic defects in each of these systems are listed in Table 1.13.
Hartnup Disease (OMIM 234500)
Hartnup disease is a rare familial condition characterized by photosensitive dermatitis, intermittent cerebellar ataxia, mental disturbances, and renal aminoaciduria. The name is that of the family in which it was first detected (262). The first gene to be identified was that for SLC6A19, a sodium-dependent amino acid transporter, on chromosome 5p15. Not all families are linked to chromosome 5p15, and there remains at least one other causative gene to be identified (263,264).
Molecular Genetics and Biochemical Pathology
The symptoms are the result of an extensive disturbance in sodium-dependent transport of neutral amino acids across the membrane of the brush border of the small intestine and the proximal renal tubular epithelium. Four main biochemical abnormalities exist: a renal aminoaciduria, increased excretion of indican, an abnormally high output of nonhydroxylated indole metabolites, and increased fecal amino acids. These deficits are discussed in detail by Milne and colleagues (265) and Scriver (266).
Pathologic Anatomy
Pathologic changes in the brain are nonspecific and are limited to neuronal degeneration and dysmyelination (267).
Clinical Manifestations
The incidence of the biochemical lesion responsible for Hartnup disease is 1 in 18,000 in Massachusetts, 1 in 70,000 in Vienna, and 1 in 33,000 in New South Wales, Australia (268). Clinical manifestations of Hartnup disease are the consequence of several factors. Polygenic inheritance is the major determinant for plasma amino acid levels, and symptoms are seen only in patients with the lowest amino acid concentrations. Because protein malnutrition further lowers amino acid levels, the disease
itself, as distinguished from its biochemical defect, is seen mainly in malnourished children. Whenever dietary intake is satisfactory, neither neurologic nor dermatologic signs appear (268). In addition, no difference exists in rate of growth or IQ scores between groups with Hartnup disease and control groups. In the series of Scriver and coworkers, 90% of Hartnup patients had normal development (269). However, low academic performance and impaired growth were seen in those patients with Hartnup disease who, for genetic reasons, tended to have the lowest plasma amino acid levels (269).
itself, as distinguished from its biochemical defect, is seen mainly in malnourished children. Whenever dietary intake is satisfactory, neither neurologic nor dermatologic signs appear (268). In addition, no difference exists in rate of growth or IQ scores between groups with Hartnup disease and control groups. In the series of Scriver and coworkers, 90% of Hartnup patients had normal development (269). However, low academic performance and impaired growth were seen in those patients with Hartnup disease who, for genetic reasons, tended to have the lowest plasma amino acid levels (269).
TABLE 1.12 Some Rarely Encountered Defects of Amino Acid Metabolism Associated with Neurologic Symptoms | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
When present, symptoms are intermittent and variable, and tend to improve with increasing age. A characteristic red, scaly rash appears on the exposed areas of the face, neck, and extensor surfaces of the extremities. This rash resembles the dermatitis of pellagra and, like it, is aggravated by sunlight. Cerebral symptoms can precede the rash for several years. They include intermittent personality changes, psychoses, migraine-like headaches, photophobia, and bouts of cerebellar ataxia. Changes in hair texture also have been observed. The four children of the original Hartnup family underwent progressive mental retardation, but this is not invariable. Renal and intestinal transport is impaired in 80% of patients and renal transport alone in 20% (269). The MRI is nonspecific; it demonstrates delayed myelination (270).
Diagnosis
Hartnup disease should be considered in patients with intermittent cerebral symptoms, even without skin involvement.
TABLE 1.13 Defects in Amino Acid Transport | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Numerous metabolic disorders with a partial enzymatic defect produce intermittent cerebellar ataxia. These include MSUD, lactic acidosis, pyruvate dehydrogenase deficiency, and some of the diseases caused by defects in the urea cycle. Additionally, episodic ataxia should be considered. This condition is caused by mutations in the calcium-channel voltage-dependent, P/Q type, alpha 1A subunit gene (CACNA1A), which is highly expressed in the cerebellum. Another rare condition to be considered in the differential diagnosis of Hartnup disease is hypertryptophanemia (see Table 1.12) and a disorder in kynurenine hydroxylation (271). Chromatography of urine for amino acids and indolic substances in the presence of a normal serum pattern is diagnostic for Hartnup disease. The absence of increased proline and hydroxyproline in the urine distinguishes Hartnup from generalized aminoaciduria that can be the result of many other disorders.
Treatment
The similarity of Hartnup disease to pellagra has prompted treatment with nicotinic acid (25 mg/day). Tryptophan ethylester also has been effective (272). However, the tendency for symptoms to remit spontaneously and for general improvement to occur with improved dietary intake and advancing age makes such therapy difficult to evaluate.
Lowe Syndrome (Oculocerebrorenal Syndrome) (OMIM 30900)
Lowe syndrome is an X-linked recessive disorder whose gene has been localized to the long arm of the X chromosome (Xq25–q26) and is characterized by severe mental retardation, myopathy, and congenital glaucoma or cataract. Biochemically, it is marked by a generalized aminoaciduria of the Fanconi type, renal tubular acidosis, and hypophosphatemic rickets (273,274). The gene responsible for the disorder has been cloned (275). It encodes a phosphatidyl inositol phosphatase located on the trans-Golgi network. The substrate for this phosphatase is a phospholipid with an important role in several basic cell processes, including cellular signaling, protein trafficking, and polymerization of the actin skeleton. Abnormalities in the structure of the actin skeleton of fibroblasts derived from patients with Lowe syndrome have been demonstrated (276). It is not clear how this lesion relates to the basic phenotypic defect, which is believed to be a defect in membrane transport (277).
Neuropathologic examination has disclosed rarefaction of the molecular layer of the cerebral cortex and parenchymal vacuolation or little more than ventricular dilation (278,279). The urinary levels of lysine are more elevated than those of the other amino acids, and defective uptake of lysine and arginine by the intestinal mucosa has been demonstrated in two patients (277).
The neurologic picture is that of a developmental delay or of progressive loss of acquired skills (280). This is accompanied by hypotonia, areflexia, and evidence of peripheral neuropathy with loss of myelinated fibers (281,282). CT scans reveal reduced density of periventricular white matter and marked scalloping of the calvarial bones, especially in the occipital region (283). T2-weighted MRI shows patchy, irregular areas of increased signal intensity
(283,285). Additionally, multiple periventricular cystic lesions have been observed (284).
(283,285). Additionally, multiple periventricular cystic lesions have been observed (284).
Heterozygous female patients are neurologically healthy, with normal renal function, but have micropunctate cortical lens opacities (285).
Creatine Transporter Defect (OMIM 300036)
This X-linked disorder was first described in 2001 by Salomons and coworkers (286). It is marked by mental retardation and hypotonia. Some patients demonstrate an extrapyramidal disorder and have a drug-resistant seizure disorder (287). The condition is not rare. Screening of European X-linked mental retardation patients identified an incidence of at least 2.1%, making it almost as common as the fragile X syndrome (FMR1 gene) (288). The defect has been localized to the creatine transporter gene (SLC6A8). Diagnosis is made by MRS, which demonstrates complete absence of the creatine peak. Urine and blood creatine levels are increased, and oral supplementation with creatine is ineffective in changing the MRS.
Creatine transporter defect should be distinguished from defects in guanidinoacetate N-methyl transferase (OMIM 601240), in which plasma guanidinoacetate levels are increased and urinary creatine levels are reduced (289). This condition is characterized by developmental arrest and deterioration, severe, early-onset seizures, and a variety of movement disorders (287,289). It should also be distinguished from arginine:glycine amidinotransferase deficiency (OMIM 602360), which presents with mental retardation, seizures, and autistic behavior and in which creatine supplementation not only normalizes the MRS, but also controls seizures and behavior (290).
DISORDERS OF CARBOHYDRATE METABOLISM AND TRANSPORT
Galactosemia (OMIM 230400)
Hepatomegaly, splenomegaly, and failure to thrive associated with the excretion of galactose were first pointed out by von Reuss in 1908 (291). Galactosemia is transmitted in an autosomal recessive manner. In the United States, it is seen with a frequency of 1 in 62,000; in Austria, 1 in 40,000 to 46,000; and in England, 1 in 72,000 (292).
Molecular Genetics and Biochemical Pathology
In 1917, Göppert demonstrated that galactosemic children excreted galactose after the ingestion of lactose (milk) and galactose (293). In 1956, Schwarz and associates found that administration of galactose to affected children gave rise to an accumulation of galactose-1-phosphate (294). This was confirmed by Kalckar and his group, who were able to demonstrate a deficiency in galactose-1-phosphate uridyltransferase (GALT), the enzyme that catalyzes the conversion of galactose-1-phosphate into galactose uridine diphosphate (UDP-galactose) (Fig. 1.8) (295). The gene for galactosemia has been mapped to the small arm of chromosome 9 (9p13). It has been cloned and sequenced, and numerous mutations have been identified (296,297). Two point mutations (Q188R and K285N) account for 69% to 80% of galactosemia in whites and result in a complete enzyme deficiency; most of the remaining mutations result in detectable amounts of enzyme activity (296,297). In African-American patients, the most common mutation (S135L) accounts for 45% of the mutant alleles. Patients homozygous for the S135L allele have residual red cell GALT activity and a milder clinical course (298). Compound heterozygotes of a classic galactosemia allele and the Los Angeles or Duarte variant are asymptomatic.
 FIGURE 1.8. Pathways of galactose metabolism. In galactosemia, galactose-1-phosphate uridyltransferase is defective. (From Ng WG, Roe TF. Disorders of carbohydrate metabolism. In: Rimoin DL, Connor JM, Pyeritz RE, et al., eds. Principles and practice of medical genetics, 4th ed. New York: Churchill Livingstone, 2002.) |
In classic galactosemia, the metabolic block is essentially complete. Lactose of human or cow’s milk is hydrolyzed to galactose and glucose, the latter being handled in a normal manner. The metabolism of galactose, however, stops after the sugar is phosphorylated to galactose-1-phosphate. This phosphate ester accumulates in erythrocytes, lens, liver, and kidney (299). Galactitol, the alcohol of galactose, also is found in the lens, brain, and urine of galactosemic individuals (300), and can be demonstrated in brain by MRS (303). Berry and coworkers postulated that galactitol induces cerebral edema that is occasionally present in neonates (301). Factors responsible for mental retardation can include a deficiency of uridine diphosphate galactose, a reduction of glycolytic intermediates, and a loss of adenosine triphosphate (ATP) (300).
Administration of galactose to affected infants results in marked hypoglycemia. This has been explained by an increased insulin release, prompted by the large amounts of circulating reducing substance, or by assuming interference by galactose-1-phosphate with normal glycogen
breakdown. The peripheral hypoglycemia is enhanced by competition between glucose and galactose at the level of the hexose transport across the blood–brain barrier. As a consequence, the brain in galactosemic patients is in a constant hypoglycemic environment.
breakdown. The peripheral hypoglycemia is enhanced by competition between glucose and galactose at the level of the hexose transport across the blood–brain barrier. As a consequence, the brain in galactosemic patients is in a constant hypoglycemic environment.
Intolerance to galactose decreases with increasing age. In part, this intolerance might be a result of the decreasing importance of milk as a food item. In patients with classic galactosemia who have been tested repeatedly, no increase in erythrocyte transferase activity has been found (302).
Pathologic Anatomy
The main pathologic lesions are found in the liver and brain. In the liver, several stages are recognized. Initially one sees a severe, diffuse, fatty metamorphosis. The hepatic cells are filled with large, pale, fat-containing vacuoles (303). If the disease remains untreated, the liver cell cords are transformed into pseudoglandular structures. The final stage is pseudolobular cirrhosis. Cerebral alterations are nonspecific. Edema, fibrous gliosis of white matter and marked loss of cortical neurons and Purkinje cells are the most prominent findings (304).
Clinical Manifestations
Infants with galactosemia appear healthy at birth, although their cord blood can already contain abnormally high concentrations of galactose-1-phosphate, and a few already have cataracts and hepatic cirrhosis, probably as a consequence of intrauterine galactosemia (305). In severe cases, symptoms develop during the first week of life. These include vomiting, diarrhea, listlessness, and failure to gain weight. Increased intracranial pressure due to cytotoxic cerebral edema can be a presenting sign (306). Infants are jaundiced. This may represent a persistence of neonatal jaundice or can appear at age 3 to 5 days (307). On a normal diet, hypoglycemia is not common. By age 2 weeks, hepatosplenomegaly and lenticular opacifications are easily detectable. The cataracts can be cortical or nuclear and can be present at birth (300). The most frequently observed opacity is a central refractile ring (308). The infants are hypotonic and often have lost the Moro reflex. Sepsis caused by Escherichia coli occurs with high frequency and is responsible for the majority of deaths during the neonatal period (309). Other secondary effects of deranged galactose metabolism include ovarian failure or atrophy and testicular atrophy (310). The MRI commonly shows multiple areas of increased signal in white matter, predominantly in the periventricular region (311).
If galactosemia goes untreated, growth failure becomes severe and the infant develops the usual signs of progressive hepatic cirrhosis. In some infants, the disease can be less severe and does not manifest until age 3 to 6 months, at which time the presenting symptoms are delayed physical and mental development. By then, cataracts can be well established and the cirrhosis far advanced.
In another group of galactosemic individuals, the diagnosis is not made until patients are several years old, often on evaluation for mental retardation. They might not have cataracts or albuminuria. Intellectual retardation is not consistent in untreated galactosemic children. When present, it is moderate; IQs range between 50 and 70. Asymptomatic homozygotes also have been detected. Most of these have some residual transferase activity.
Diagnosis
The enzyme defect is best documented by measuring the erythrocyte GALT activity. Several methods are available and have been be used for statewide and nationwide screening. In the Austrian screening program reviewed by Item and coworkers, only 1 in 190 infants who tested positive initially actually had galactosemia (297). Some of these were galactosemia carriers or Duarte/galactosemia compound heterozygotes.
Galactosuria, usually in combination with glucosuria or fructosuria, is seen in severe hepatic disorders of the neonatal period (e.g., neonatal hepatitis, tyrosinosis, congenital atresia of the bile ducts). Families in which several members are mentally defective and have congenital cataracts without an abnormality in galactose metabolism have been described by Franceschetti and others (312).
The antenatal diagnosis of galactosemia can be made by assay of GALT activity on cultured amniocytes or chorionic villi (315).
Treatment
When milk is withdrawn and lactose-free products such as Nutramigen or Prosobee are substituted, gastrointestinal symptoms are rapidly relieved and normal growth resumes. The progression of cirrhosis is arrested, and in 35% of patients, the cataracts disappear (307). Because infants have developed hypoglycemia when first placed on a galactose-free diet, it might be useful to add some glucose to the formula.
The propensity of galactosemic neonates for E. coli sepsis requires securing cultures of blood, urine, and CSF and treating against this organism until the test results are negative (309). In the larger series of Waggoner and coworkers, sepsis was suspected in 30% of neonates and confirmed in 10% (314).
Recommendations for the management of infants and children with galactosemia have been published by Walter and coworkers (315). Maintenance of the galactose-free diet and avoidance of milk and milk products is recommended until at least after puberty. In several children who returned to a milk-containing diet before puberty, cataracts flared up. Even after that age, some continue to be sensitive to milk products and prefer to avoid them. Intermittent monitoring of erythrocyte galactose-1-phosphate levels has been suggested. Even
children with well-controlled galactosemia have elevated galactose-1-phosphate levels. Endogenous formation of galactose-1-phosphate from glucose-1-phosphate by way of the epimerase reaction (see Fig. 1.8) is believed to be responsible.
children with well-controlled galactosemia have elevated galactose-1-phosphate levels. Endogenous formation of galactose-1-phosphate from glucose-1-phosphate by way of the epimerase reaction (see Fig. 1.8) is believed to be responsible.
The long-term outlook for galactosemic patients is not as good as was initially believed, even when the diet is carefully monitored. A number of cases of progressive cerebellar ataxia and extrapyramidal movement disorders have been reported (316,317). In the series of Waggoner and colleagues, cerebellar deficits were seen in 18% (314); in the series of Kaufman and colleagues, the incidence was 27% (317). The cause for this syndrome is unclear, and MRI studies on patients with this syndrome do not differ from those without it (317). Rogers and Segal postulated that the syndrome results from an endogenous product of galactose-1-phosphate from UDP-glucose via the epimerase reaction (318). Another possibility is that a deficiency of UDP-galactose could limit the formation of cerebral glycoproteins and galactolipids. Administration of uridine, which has been suggested to prevent these complications, has not been effective (319).
Cognitive deficits are common. In 70% of galactosemic children treated from birth, the IQ was 90 or higher; however, approximately 50% of the youngsters had significant visual and perceptual deficits, and 33% had EEG abnormalities (320). Studies confirm that most patients have cognitive deficits in one or more areas. Verbal dyspraxia occurs in some 62%. Patients are unable to program their speech musculature and also show frequent disturbances in speech rhythm. Receptive language is normal (317,321). In addition, there appears to be a progressive decline in IQ with age (314,317). Neither IQ scores nor the presence of the dyspractic speech disorder are highly related with the age at which therapy is initiated or quality of control (314). Instead, cognitive and language deficits are likely to result from the in utero formation of potentially neurotoxic galactose-1-phosphate and the continued generation of galactose from glucose via UDP-galactose-4-epimerase (see Fig. 1.8) (300,322).
Two other defects of galactose metabolism have been recognized. A deficiency of galactokinase (see Fig. 1.8) is the more common (OMIM 230200). Cataracts are present in most patients and pseudotumor cerebri has been seen occasionally. The outlook in terms of mental function is better than for patients with galactosemia, and most appear to be normal (323). A very rare disorder, generalized deficiency of epimerase (OMIM 230350), the enzyme that converts UDP-glucose to UDP-galactose (see Fig. 1.8), can result in galactosuria, failure to thrive, sensorineural deafness, dysmorphic features, and mental retardation (324).
Fructose Intolerance (OMIM 229600)
Fructose intolerance, as distinguished from benign fructosuria, was first described by Chambers and Pratt in 1956 (325). The condition is transmitted as an autosomal recessive trait and is the consequence of a deficiency in the principal hepatic aldolase, aldolase B (326). The gene coding for aldolase B is located on the long arm of chromosome 9 (9q21.3–q22.2), and more than 25 mutations have been documented (327).
As a consequence of the metabolic defect, ingested fructose (or sucrose, which is split into fructose and glucose) is converted to fructose-1-phosphate, which accumulates in tissues and is responsible for renal and hepatic damage. Additionally, plasma lactate and urate levels increase (328). A glucagon-unresponsive hypoglycemia results from the blockage of glycogenolysis by fructose-1-phosphate at the point of phosphorylase and from the interruption of gluconeogenesis by the phosphate ester at the level of the mutant fructose-1,6-diphosphate aldolase.
The main pathologic abnormality is hepatic cirrhosis similar to that seen in galactosemia. The brain shows retarded myelination and neuronal shrinkage attributable to hypoglycemia (329). An associated coagulation defect can induce intracerebral hemorrhage.
Hereditary fructose intolerance is relatively rare in the United States and far more common in Europe. In Switzerland, the gene frequency is 1 in 80, and in Great Britain, 1 in 250 (328). The condition manifests by intestinal disturbances, poor weight gain, and attacks of hypoglycemia after fructose ingestion. Transient icterus, hepatic enlargement, fructosuria, albuminuria, and aminoaciduria follow intake of large quantities of fructose. Mild mental deficiency is frequent, and there may be a flaccid quadriparesis (330). Heterozygotes are predisposed to gout (328).
Diagnosis is based in part on the patient’s clinical history, on the presence of a urinary reducing substance, and on the results of an intravenous fructose tolerance test (0.25 g/kg), a procedure that should be performed carefully under monitored conditions. An oral tolerance test is contraindicated in view of the ensuing severe gastrointestinal and systemic symptoms (331). For confirmation, a jejunal or liver biopsy with determination of fructose-1-phosphate aldolase levels is necessary (332). MRS has been used to confirm the diagnosis and to determine heterozygosity (328).
Other causes of increased fructose excretion include impaired liver function and fructosuria, which is an asymptomatic metabolic variant caused by a deficiency of fructokinase (OMIM 299800).
Treatment of hereditary fructose intolerance is relatively simple but is needed lifelong. It involves avoiding the intake of fruits and cane or beet sugar (sucrose).
Other Disorders of Carbohydrate Metabolism
Fructose-1,6-diphosphatase deficiency (OMIM 229700) is a rare disorder that manifests during the neonatal period
with hyperventilation, hyperbilirubinemia, seizures, coma, and laboratory evidence of ketosis, hypoglycemia, and lactic acidosis (333). The diagnosis is difficult, and a variety of other causes for intermittent neonatal hypoglycemia must be excluded. The enzyme defect can be demonstrated by liver biopsy (334).
with hyperventilation, hyperbilirubinemia, seizures, coma, and laboratory evidence of ketosis, hypoglycemia, and lactic acidosis (333). The diagnosis is difficult, and a variety of other causes for intermittent neonatal hypoglycemia must be excluded. The enzyme defect can be demonstrated by liver biopsy (334).
Of the various forms of mellituria seen in infancy and childhood, the most common are caused by the increased excretion of a single sugar, predominantly glucose. Isolated lactosuria and fructosuria are encountered also. Lactosuria usually is explained on the basis of congenital lactose intolerance or secondary lactose intolerance associated with enteritis, celiac disease, and cystic fibrosis. Essential pentosuria, one of the original inborn errors of metabolism described by Garrod, is a result of the excretion of L-xylulose. Ribosuria occurs in Duchenne muscular dystrophy, probably as the result of tissue breakdown. Sucrosuria has been reported in association with hiatal hernia and other intestinal disturbances.
A mixed-sugar excretion also can be seen in acute infections, liver disease, and gastroenteritis.
Glucose Transporter 1 Deficiency Syndrome (De Vivo Disease) (OMIM 606777)
This condition was first described in 1991 by De Vivo and coworkers (335). It is transmitted as an autosomal dominant trait, and, as the name indicates, results from a mutation in the gene for the glucose 1 transporter (GLUT 1). De Vivo and coworkers reviewed the molecular genetics of this condition (336).
The clinical picture is marked by the early onset of a seizure disorder accompanied by delayed development, the acquisition of microcephaly, incoordination, and spasticity. Other paroxysmal events have been observed. These include intermittent ataxia, confusion, or somnolence. Patients with intermittent ataxia and mental retardation in the absence of seizures have been reported, as have opsoclonic eye movements and a complex motor disorder with elements of ataxia, dystonia, and spasticity (336,337).
The presence of hypoglycorrhachia in the presence of normal blood sugar is diagnostic. Neuroimaging studies are generally unremarkable. In about 50% of patients the EEG demonstrates generalized spike or polyspike and wave discharges. Diffuse or focal slowing has also been observed. An erythrocyte glucose uptake study will confirm the diagnosis (338).
Treatment by means of the ketogenic diet or medium-chain triglycerides has been fairly effective. Ketone bodies are transported across the blood–brain barrier by monocarboxylic transporters and can be used as an alternative brain fuel. Barbiturates and caffeine, which inhibit glucose transport across the blood–brain barrier, should be avoided. α-Lipoic acid supplementation may be beneficial (336).
ORGANIC ACIDURIAS
A number of disorders of intermediary metabolism are manifested by intermittent episodes of vomiting, lethargy, acidosis, and the excretion of large amounts of organic acids. Even though the enzymatic lesions responsible are not related, they are grouped together because they are detected by analysis of urinary organic acids.
Propionic Acidemia (Ketotic Hyperglycinemia) (OMIM 232000; 232050)
Propionic acidemia, the first of the organic acidurias to be described, is characterized by intermittent episodes of vomiting, lethargy, and ketosis. Hsia and coworkers demonstrated a defect in propionyl-CoA carboxylase, a biotin-dependent enzyme that converts propionyl-CoA to methylmalonyl-CoA (339). The enzyme consists of two polypeptides, α and β, coded by genes (PCCA and PCCB) that are located on chromosomes 13 and 3, respectively. Considerable genetic heterogeneity exists, with defects in each of the two structural genes encoding the two subunits of propionyl-CoA carboxylase. Most of these are single-base substitutions (340). The clinical presentation of the two forms appears to be comparable; the original family had the PCCA type of propionic acidemia.
As a consequence of the metabolic block, not only is serum propionate elevated, but also several propionate derivatives accumulate (341). Hyperammonemia is frequent, probably as a consequence of an inhibition by the accumulating organic acids of N-acetylglutamate synthetase, the enzyme that forms N-acetylglutamate, a stimulator of carbamoyl-phosphate synthetase (see Fig. 1.6) (342). The severity of hyperammonemia appears to be proportional to the serum propionate levels (343). The mechanism for hyperglycinemia in this and in several of the other organic acidurias has not been fully elucidated. It appears likely, however, that propionate interferes with one of the components of the mitochondrial glycine-cleavage system.
In the classic form of the disease, symptoms start shortly after birth (344). In other cases, they might not become apparent until late infancy or childhood and are precipitated by upper respiratory or gastrointestinal infections. Marked intellectual retardation and a neurologic picture of a mixed pyramidal and extrapyramidal lesion ultimately become apparent (345). A less severe form of the disease is fairly common in Japan. It presents with mild mental retardation and extrapyramidal symptoms. It results from a mutation in PCCB (346). As a rule, attacks are precipitated by ingestion of proteins and various amino acids, notably leucine. In addition to ketoacidosis, hyperammonemia, persistent neutropenia, and thrombocytopenia occur. Propionic acidemia is seen in all patients, and plasma and urinary glycine levels are increased
(347). In some patients, MRI shows increased signal in the caudate nucleus and putamen on T2-weighted images (348).
(347). In some patients, MRI shows increased signal in the caudate nucleus and putamen on T2-weighted images (348).
Structural abnormalities in the brain are similar to those seen in PKU and other diseases of amino acid metabolism, namely retarded myelination and a spongy degeneration of gray and white matter (349,350).
Treatment involves a diet low in valine, isoleucine, threonine, and methionine, with carnitine supplementation (60 to 200 mg/kg per day). A commercial formula is available. A few patients have responded to biotin. Patients generally require a gastrostomy tube because of poor feeding. Courses of metronidazole have been used to decrease the amount of propionate produced by anaerobic intestinal bacteria. Cardiomyopathy and arrhythmias can further complicate the disease.
Propionic acidemia is biochemically and clinically distinct from familial glycinuria (OMIM 138500), a condition in which serum glycine levels are normal and the nervous system is unaffected (351). It also should be distinguished from iminoglycinuria and from nonketotic hyperglycinemia. Isovaleric acidemia and α-ketothiolase deficiency also can present with episodes of ketoacidosis and hyperglycinemia.
Methylmalonic Aciduria (OMIM 251000)
The classic presentation of MMA is one of acute neonatal ketoacidosis, with lethargy, vomiting, and profound hypotonia. Several genetic entities have been recognized. In all, the conversion of methylmalonyl-CoA to succinyl-CoA is impaired, a result either of a defect in the mitochondrial apoenzyme, methylmalonyl-CoA mutase, or in the biosynthesis, transport, or absorption of its adenosylcobalamin cofactor (352). The gene for methylmalonyl-CoA mutase has been cloned, and more than two dozen mutations have been recognized (353).
Approximately one-third of patients with MMA have the classic form of the disease (352). In this condition, the defect is localized to the apoenzyme methylmalonyl-CoA mutase. In the majority of patients, the enzyme is totally inactive, whereas in other patients with MMA, the apoenzyme defect is partial (354). In the remaining patients with MMA, constituting approximately 50% of a series of 45 patients assembled by Matsui and her group (352), dramatic biochemical improvement occurs with the administration of adenosylcobalamin, the cofactor for the mutase. In most of the responders, synthesis of adenosylcobalamin is blocked at the formation of the cofactor from cobalamin (cobalamin adenosyltransferase deficiency) or at one of the mitochondrial cobalamin reductases. In a small proportion of patients, the defect appears to involve adenosylcobalamin and methylcobalamin. Because the latter serves as cofactor for the conversion of homocysteine to methionine, children with this particular defect demonstrate homocystinuria and MMA (355). The various disorders in cobalamin absorption, transport, and use were reviewed by Shevell and Rosenblatt (356).
As ascertained by routine screening of newborns, a large proportion of infants with persistent MMA are asymptomatic and experience normal growth and mental development (357). These children may represent the mildest form of partial mutase deficiency. Small elevations in MMA and homocystine are also seen with maternal vitamin B12 deficiency, particularly common in vegans.
Infants suffering from the classic form of the disease (absence of the apoenzyme methylmalonyl-CoA mutase) become symptomatic during the first week of life, usually after the onset of protein feedings. The clinical picture is highlighted by hypotonia, lethargy, recurrent vomiting, and profound metabolic acidosis (352). Survivors of the initial crisis have recurrent episodes with intercurrent illnesses and often have spastic quadriparesis, dystonia, and severe developmental delay. When the apoenzyme defect is partial, patients generally become symptomatic in late infancy or childhood and are not as severely affected (354). MRI studies of the brain resemble those obtained on patients with propionic acidemia with delayed myelination and changes in the basal ganglia (358).
Pathologic changes within the brain also involve the basal ganglia predominantly, with neuronal loss and gliosis, or spongy changes in the globus pallidus and putamen. Abnormal laboratory findings in the classic form of MMA include ketoacidosis and an increased amount of methylmalonic acid in blood and urine. Hyperglycinemia is seen in 70% of classic cases, and hyperammonemia is seen in 75% (352). Hematologic abnormalities, notably leukopenia, anemia, and thrombocytopenia, are encountered in approximately 50% of the cases. These abnormalities result from the growth inhibition of marrow stem cells by methylmalonic acid (359).
Almost all of the survivors have some degree of neurologic impairment. This is in part the consequence of diminished protein tolerance and frequent bouts of metabolic decompensation. MMA is nephrotoxic leading to renal failure in the second decade of life (360). Late-onset patients fare better, and some have relatively minor neuromotor and mental handicaps (361).
At least four disorders of cobalamin absorption and transport and seven disorders of cobalamin utilization exist. In two of the cobalamin utilization defects, the output of methylmalonic acid is increased (cblA, cblB); in three, MMA and homocystinuria occur (cblC, cblD, and cblF); and in two, there is an increased excretion of homocystine (cblE and cblG) (356).
The clinical course of children experiencing a cofactor deficiency is not as severe as that of children with an apoenzyme deficiency, and nearly 60% of patients with the most common cobalamin utilization defect, cobalamin reductase deficiency (cblC), do not become symptomatic until after age 1 month (352). The clinical picture is variable (362). The majority of children present in infancy or
during the first year of life with failure to thrive, developmental delay, and megaloblastic anemia. In others, the disease develops during adolescence, with dementia and a myelopathy (363,364). A progressive retinal degeneration has been observed (365). Pathologic findings in the cblC defect include a subacute combined degeneration of the spinal cord and a thrombotic microangiopathy (366).
during the first year of life with failure to thrive, developmental delay, and megaloblastic anemia. In others, the disease develops during adolescence, with dementia and a myelopathy (363,364). A progressive retinal degeneration has been observed (365). Pathologic findings in the cblC defect include a subacute combined degeneration of the spinal cord and a thrombotic microangiopathy (366).
The diagnosis of MMA can be suspected when urine treated with diazotized p-nitroaniline turns emerald green. MMA also is seen in pernicious anemia and vitamin B12 deficiency and in the infantile form of mitochondrial depletion syndrome (see Chapter 2). Cultured fibroblasts are used for the further delineation of the various metabolic defects responsible for MMA.
Infants with MMA should first be tested for vitamin B12 responsiveness (1 to 2 mg of cyanocobalamin or preferably hydroxycobalamin intramuscularly daily for several days). In those who fail to respond, a low-protein diet (0.75 to 1.2 g protein/kg per day) with the addition of L-carnitine is required (367). Vitamin B12-responsive patients are treated with oral (1 mg/day) or intramuscular cobalamin (1 mg every 3 weeks), supplemented with L-carnitine (50 to 200 mg/kg per day) (369). In the child who does not respond to hydroxycobalamin, a trial of deoxyadenosylcobalamin is indicated, but the substance is hard to obtain (367). In children with the various cofactor deficiencies, the biochemical and clinical response to therapy is gratifying (362,368). Even an occasional patient with the apoenzyme deficiency can improve with therapy. Hepatic transplantation has been used in some of the more severe cases of vitamin B12-nonresponsive methylmalonic aciduria. It appears that the procedure does not prevent neurologic dysfunction or progressive renal failure (369).
Isovaleric Aciduria (OMIM 243500)
A striking odor of urine, perspiration, and exhaled air, resembling stale perspiration, is characteristic of patients with isovaleric aciduria. The enzymatic lesion in this condition has been localized to isovaleryl-CoA dehydrogenase (see Fig. 1.5, step 3), a mitochondrial enzyme (370). The gene for this enzyme has been mapped to the long arm of chromosome 15 (15q12–q15) and has been cloned (371). As a consequence of the enzymatic defect, serum isovaleric acid concentrations are several hundred times normal, and the administration of L-leucine produces a sustained increase in isovaleric acid levels.
In addition to abnormally elevated concentrations of isovaleric acid in blood and urine, moderate hyperammonemia occurs. This is particularly evident during the neonatal period. Large quantities of isovaleryl glycine are excreted during acute episodes of acidosis and while the patient is in remission. During attacks, 3-hydroxyisovaleric acid, 4-hydroxyisovaleric acid and its oxidation products (methylsuccinic acid and methaconic acids), and isovaleryl glucuronide are excreted also (372). The acidosis seen during an attack appears to result from an accumulation of ketone bodies rather than from the presence of isovaleric acid.
Two clinical phenotypes are seen: an acute and commonly fatal neonatal form in which infants develop recurrent acidosis and coma during the first week of life, and a chronic form with recurrent attacks of vomiting, lethargy, ataxia, and ketoacidosis (373). Acute and chronic forms can be present in the same family. No correlation exists between the amount of residual enzyme activity and the clinical form of the disease. Instead, the severity of the disease appears to be a function of the effectiveness with which isovaleryl-CoA is detoxified to its glycine derivative (374). Attacks are triggered by infections or excessive protein intake. Pancytopenia is not uncommon; it is caused by arrested maturation of hematopoietic precursors (375).
Treatment involves a low-protein diet (1.5 to 2 g/kg per day) with L-carnitine (50 to 100 mg/kg per day). Glycine (250 mg/kg per day) has been used instead of carnitine. Glycine and carnitine aid in the mitochondrial detoxification of isovaleryl-CoA (374). For infants who survive the neonatal period, the outlook for normal intellectual development is fairly good, and 4 of 9 patients treated by Berry and coworkers at Children’s Hospital of Philadelphia had IQs of greater than 95 (374).
A clinical picture resembling isovaleric aciduria develops after intoxication with achee, the fruit that induces Jamaican vomiting sickness and that contains hypoglycin, an inhibitor of several acyl-CoA dehydrogenases, including isovaleryl-CoA dehydrogenase (376).
Glutaric Acidurias
Several completely different genetic defects have been grouped under the term glutaric aciduria.
Glutaric Aciduria Type I (OMIM 231670)
This is a recessive disorder with an incidence of approximately 1 in 30,000. It is caused by a defect in the gene for glutaryl-CoA dehydrogenase. Biochemically, the condition is marked by the excretion of large amounts of glutaric, 3-hydroxyglutaric, and glutaconic acids. The clinical picture is protean, and the disorder is frequently undiagnosed or misdiagnosed. In approximately 20% of patients, glutaric aciduria I takes the form of a neurodegenerative condition commencing during the latter part of the first year of life and characterized by hypotonia, dystonia, choreoathetosis, and seizures (377). In most of the remaining patients, development is normal for as long as 2 years of age, and then, after what appears to be an infectious, an encephalitic, or a Reye syndrome–like illness or following routine immunization, neurologic deterioration occurs (378,379). In yet other cases, the clinical picture
resembles extrapyramidal cerebral palsy (380). Rare cases remain asymptomatic. Macrocephaly is noted in 70% of cases (381). Lack of appetite, sleeplessness, and profuse sweating also is noted, as is hypoglycemia (382,383). No molecular basis for the clinical variability exists, and the severity of the clinical phenotype seems to be closely linked to the development of encephalopathic crises rather than to residual enzyme activity or genotype (384).
resembles extrapyramidal cerebral palsy (380). Rare cases remain asymptomatic. Macrocephaly is noted in 70% of cases (381). Lack of appetite, sleeplessness, and profuse sweating also is noted, as is hypoglycemia (382,383). No molecular basis for the clinical variability exists, and the severity of the clinical phenotype seems to be closely linked to the development of encephalopathic crises rather than to residual enzyme activity or genotype (384).
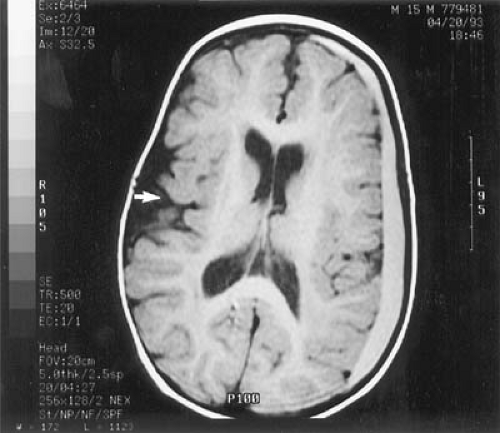 FIGURE 1.9. Glutaric aciduria I. Magnetic resonance imaging study. This T1-weighted image demonstrates an enlarged operculum on the right (arrow) and an extensive left subdural hematoma. |
Neuroimaging shows frontotemporal cortical atrophy giving what has been termed a bat wing appearance. This is often accompanied by increased signal in the basal ganglia on T2-weighted images and caudate atrophy. Bilateral subdural hematomas have also been described (Fig. 1.9). When these are accompanied by retinal hemorrhages, an erroneous diagnosis of child abuse frequently is made (390).
Neuropathology shows temporal and frontal lobe hypoplasia, degeneration of the putamen and globus pallidus, mild status spongiosus of white matter, and heterotopic neurons in cerebellum (385). It is not clear why so much of the damage is localized to the basal ganglia. Kolker and coworkers postulated that 3-hydroxyglutaric acid, which is structurally related to glutamic acid, induces excitotoxic cell damage, and that in addition 3-hydroxyglutaric acid and glutaric acid modulate glutamatergic and gamma-aminobutyric acid (GABA)-ergic neurotransmission. Secondary amplification loops could also potentiate the neurotoxic properties of these organic acids (386).
A low-protein, high-calorie diet supplemented with a formula lacking lysine and tryptophan and containing carnitine has sometimes prevented further deterioration, but in the Scandinavian series of Kyllerman and colleagues almost all patients were left with a severe dystonic-dyskinetic disorder (387). The experience of Hoffmann and coworkers was similar (381). Strauss and colleagues, who worked with the disease in the Pennsylvania Amish population, however, found that good dietary care can reduce the incidence of basal ganglia injury to 35% (388).
Glutaric Aciduria Type II (Multiple Acyl-CoA Dehydrogenase Deficiency) (MADD) (OMIM 231680)
In this condition the defect has been localized to one of three genes involved in the mitochondrial β-oxidation of fatty acids: those coding for the α and β subunits of the electron transfer flavoprotein (ETF) and that coding for the electron transfer flavoprotein dehydrogenase (389,390). The clinical picture of GA II due to the different defects appears to be nondistinguishable; each defect can lead to a range of mild or severe cases, depending presumably on the location and nature of the intragenic lesion, that is, mutation, in each case. As a consequence of the enzyme deficiency there is increased excretion not only of glutaric acid, but also of other organic acids, including lactic, ethylmalonic, isobutyric, and isovaleric acids.
The heterogeneous clinical features of patients with MADD fall into three classes (391): a neonatal-onset form with congenital anomalies (type I), a neonatal-onset form without congenital anomalies (type II), and a late-onset form (type III).
Glutaric aciduria II can present in the neonatal period with an overwhelming and generally fatal metabolic acidosis coupled with hypoglycemia, acidemia, and a cardiomyopathy. Affected infants can have an odor of sweaty feet. As a rule, there is good correlation between the severity of the metabolic block, rather than its location and the severity of the disease, with null mutations producing the development of congenital anomalies and the presence of a minute amount of enzymatic activity allowing the development of type II disease (392).
Dysmorphic features are prominent in approximately one-half of cases (393). They include macrocephaly, a large anterior fontanel, a high forehead, a flat nasal bridge, and malformed ears. This appearance is reminiscent of Zellweger syndrome (Fig. 1.10). Neuroimaging discloses agenesis of the cerebellar vermis and hypoplastic temporal lobes (394). The neonatal-onset forms are usually fatal and are characterized by severe nonketotic hypoglycemia, metabolic acidosis, multisystem involvement, and excretion of large amounts of fatty acid- and amino acid-derived metabolites.
Standard treatment consists of a high-carbohydrate, low-fat and protein diet supplemented with carnitine. Van Hove and coworkers suggested that this condition be treated with D,L-3-hydroxybutyrate (430 to 700 mg/kg
per day). In their experience this treatment resulted in improvement of neurologic function and cardiac contractility (395).
per day). In their experience this treatment resulted in improvement of neurologic function and cardiac contractility (395).
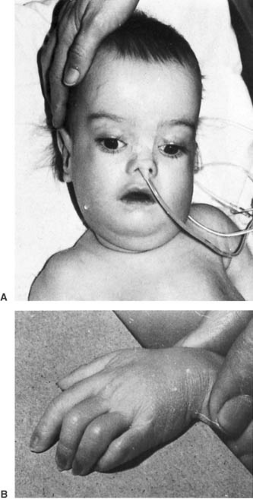 FIGURE 1.10. Zellweger syndrome. A: Typical facies with high forehead and flat facies. Redundant skin of the neck is seen. (Courtesy of the late Dr. H. Zellweger, University of Iowa, Iowa City, IA.) B: Hand, demonstrating camptodactyly of third, fourth, and fifth fingers. (Courtesy of Dr. J. M. Opitz, Shodair Children’s Hospital, Helena, MT.) |
Symptoms and age at presentation of late-onset MADD (type III) are highly variable and characterized by recurrent episodes of lethargy, vomiting, hypoglycemia, metabolic acidosis, and hepatomegaly often preceded by metabolic stress. Muscle involvement in the form of pain, weakness, and lipid storage myopathy also occurs. The organic aciduria in patients with the late-onset form of MADD is often intermittent and only evident during periods of illness or catabolic stress. Other cases present in early childhood with progressive spastic ataxia and high signal intensity on T2-weighted MRI in supratentorial white matter, a picture mimicking a leukodystrophy (398).
The neuropathologic picture is marked by diffuse gliosis of cerebrum, brainstem, and cerebellum, with foci of leukomalacia and striatal degeneration (393,397).
Glutaric Aciduria Type III (OMIM 231690)
This condition is the result of a defect of peroxisomal glutaryl CoA oxidase. It results in a persistent isolated glutaric acid excretion, but the phenotype of this disorder is unknown, and it may well represent a benign inborn error (400).
The other, rarer organic acidurias are summarized in Table 1.15.
DISORDERS OF FATTY ACID OXIDATION
Up to 18 defects of hepatic mitochondrial fatty acid oxidation have been recognized, including eight defects of the β-oxidation cycle (401,402). Affected patients are unable to use fatty acids derived from adipose tissue or diet for energy production or hepatic ketone synthesis. Because fatty acids are the principal energy source during fasting, infants rapidly decompensate in the neonatal period or during a febrile illness. In addition to the acute metabolic decompensation that results when infants are fasted, resulting in a rapid evolution of seizures and coma, the clinical presentation of most of these conditions is marked by episodes of a nonketotic or hypoketotic hypoglycemia. The esterified-to-free-carnitine ratio is increased, and generally there is hypocarnitinemia. When the enzyme defect is limited to muscle, it can present with cardiomyopathy, muscle weakness, and myoglobinuria. These symptoms are features of defects in the transport of fatty acids into mitochondria and are covered in Chapter 2. Disorders of fatty acid oxidation are uncommon, and collectively, their incidence in Great Britain has been estimated at 1 in 5,000 live births (403).
Medium-Chain Acyl-CoA Dehydrogenase (MCAD) Deficiency (OMIM 201450)
The most common of the disorders of fatty acid oxidation is a defect involving medium-chain acyl-CoA dehydrogenase (MCAD). The incidence of this entity varies among
population groups. In whites, it has been estimated to be between 1 in 6,400 and 1 in 23,000 live births; thus, it appears to be almost as common as PKU (404,405,406).
population groups. In whites, it has been estimated to be between 1 in 6,400 and 1 in 23,000 live births; thus, it appears to be almost as common as PKU (404,405,406).
TABLE 1.14 Fatty Acid Oxidation Disorders | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
Because MCAD is active over the range of C4 to C12 carbons, its deficiency permits fatty acid oxidation to progress only up to the point at which the carbon chain has been reduced to 12 (402). A single point mutation in the gene is responsible for the condition in more than 90% of symptomatic white patients. This mutation results in the substitution of glutamine for lysine in the MCAD precursor. This disrupts folding of the mature form and its assembly in mitochondria, with subsequent disappearance of the mutant MCAD.
Clinical symptoms usually develop during the first year of life and are characterized by episodes of hypoketotic hypoglycemia triggered by fasting or infections. These episodes result in lethargy, vomiting, and altered consciousness. In the experience of Rinaldo and colleagues, mortality of the first such episode of metabolic decompensation is 59% (407). In the series of Iafolla and coworkers, 36% of patients had sudden cardiorespiratory arrest (404). Although the last presentation resembles that of sudden infant death syndrome (SIDS), Miller and colleagues were unable to find any homozygotes for the glutamine-to-lysine mutation leading to MCAD deficiency in 67 SIDS babies (408), and Boles and colleagues found an incidence of only 0.6% among cases diagnosed as SIDS (409). In the latter study, another 0.6% of SIDS cases were believed to have glutaric aciduria II (409). The general practice of most coroners is to evaluate all cases of SIDS for an underlying metabolic disorder using blood, bile, vitreous humor, or tissue.
The MCAD deficiency is treated by supplemental L-carnitine, a low-fat diet, and avoidance of fasting, which rapidly initiates a potentially fatal hypoglycemia, which must be corrected by the prompt institution of glucose. The efficacy of supplemental L-carnitine is unclear. On follow-up of symptomatic patients, a significant proportion of survivors have global developmental delays, attention deficit disorder, and language deficits (404). Detection by tandem mass spectrometry newborn screening and early treatment can prevent this outcome.
Other Disorders of Fatty Acid Oxidation
Very Long Chain Acyl-CoA Deficiency (VLCAD) (OMIM 201475)
The other disorders of fatty acid β-oxidation are much less common. They are summarized in Table 1.14. In VLCAD the defect is at the first step of the fatty acid β-oxidation cycle. Clinically, failure of long-chain fatty acid β-oxidation leads to hypoketotic hypoglycemia associated with coma, liver dysfunction, skeletal myopathy, and hypertrophic cardiomyopathy. Patients are unable to metabolize fatty acids with 12 to 18 carbons (420,421,422,423). Three clinical forms have been recognized: a severe form with early onset and a high incidence of cardiomyopathy; a milder, childhood-onset form; and an adult form with rhabdomyolysis and myoglobinuria (422,423,424). The clinical picture appears to correlate with the amount of residual enzyme activity (417). VLCAD can be effectively treated with avoidance of fasting and a diet low in long-chain fats supplemented with medium-chain triglyceride oil and carnitine. Uncooked cornstarch at night can be added to the regimen (425).
Short-Chain Acyl-CoA Dehydrogenase Deficiency (SCAD) (OMIM 201470)
The short-chain acyl dehydrogenase is active from four to six carbons, and SCAD infants excrete large amounts of butyrate, ethylmalonate, methylmalonate, and methylsuccinate (426,427). The clinical picture is complex. Some patients develop acute acidosis and muscle weakness early in life; others develop a multicore myopathy in adolescence or in their adult years, and yet others remain clinically well (427,428). Whereas patients with MCAD and long-chain acyl-CoA deficiency tend to develop hypoketotic hypoglycemia, patients with short-chain acyl-CoA dehydrogenase deficiency may not have hypoglycemia. Most symptomatic patients have hypotonia, hyperactivity, and
developmental delay. The condition can be intermittently symptomatic. In cases detected by newborn screening, care must be taken to determine whether the child is truly deficient and thus potentially at risk or merely harbors a common polymorphism in SCAD (429,430).
developmental delay. The condition can be intermittently symptomatic. In cases detected by newborn screening, care must be taken to determine whether the child is truly deficient and thus potentially at risk or merely harbors a common polymorphism in SCAD (429,430).
Long-Chain Hydroxy Acyl Co-A Deficiency (LCHAD) (OMIM 143450)
The mitochondrial trifunctional protein (MTP) is a multienzyme complex that catalyzes three of the four chain-shortening reactions in the β-oxidation of long-chain fatty acids. It is composed of four alpha subunits harboring long-chain enoyl-CoA hydratase and long-chain L-3-hydroxyacyl-CoA dehydrogenase and four beta subunits carrying the long-chain 3-ketoacyl-CoA dehydrogenase. Mutation in either subunit can result in reduced activity of all three enzymes.
Different phenotypes have been reported. There is a severe and generally fatal neonatal presentation with cardiomyopathy, Reye-like symptoms; a hepatic form with recurrent hypoketotic hypoglycemia; and a late-onset myopathic form with recurrent myoglobinuria (419,430a).
Defects in branched-chain acyl-CoA dehydrogenation also have been recognized. They are listed in Table 1.15.
The diagnosis of these disorders depends on the presence of the various dicarboxylic acids in urine, and more recently by the application of tandem mass spectrometry. The pros and cons of expanded neonatal screening to include the disorders of fatty acid oxidation are discussed by Dezateux (431).
Disorders of Biotin Metabolism
Four biotin-dependent enzymes have been described. All are carboxylases: propionyl-CoA carboxylase, 3-methylcrontonyl-CoA carboxylase, pyruvate carboxylase, and acetyl-CoA carboxylase. The covalent binding of biotin to these enzymes is catalyzed by holocarboxylase synthetase. A significant proportion of infants with organic aciduria are found to have impaired function of all four of these biotin-containing carboxylases (multiple carboxylase deficiency, holocarboxylase synthetase deficiency, OMIM 253270) (432). Most patients diagnosed with this condition became symptomatic in early infancy with metabolic acidosis, ketosis, and an erythematous rash. High concentrations of the metabolites α-hydroxyisovalerate, α-methylcrotonylglycine, α-hydroxypropionate, methyl citrate, and lactate are excreted in urine, which acquires a distinctive odor. The condition is caused by a deficiency in biotin holocarboxylase synthetase that is never complete (433). Symptoms are usually reversed by biotin, given in doses between 10 and 80 mg/day (434), but any permanent damage remains.
A second, more common disorder of biotin metabolism results from a deficiency of biotinidase (biotinidase deficiency, OMIM 253260). This condition is characterized by the onset of symptoms after the neonatal period, usually between ages 2 and 3 months. Biotinidase hydrolyzes the bond between biotin and lysine, the bound form in which biotin exists in the diet, and thus recycles biotin in the body (435). The enzyme deficiency can be complete or partial, with patients who have a partial deficiency tending to be asymptomatic unless stressed by prolonged infections (436). Considerable heterogeneity exists in profound biotinidase deficiency, and numerous mutations have been recognized (437). As ascertained by a screening program, the incidence of complete biotinidase deficiency in the United States is 1 in 166,000. The condition is rare in Asians (438).
Symptoms in complete biotinidase deficiency include lactic acidosis, alopecia, ataxia, spastic paraparesis, seizures, and an erythematous rash. Developmental delay soon becomes apparent. Hearing loss, acute vision loss, optic atrophy, and respiratory irregularities are seen in a significant proportion of cases (432,434). Plasma and urinary biotin levels are subnormal, and clinical and biochemical findings are rapidly reversed by the administration of oral biotin (5 to 10 mg/kg per day) (434). Children treated presymptomatically remain normal; those treated after becoming symptomatic are left with residual deficits, hearing impairment, optic atrophy, and developmental delay (439).
A unique familial syndrome of acute encephalopathy appearing between 1 and 14 years of age and marked by confusion and lethargy progressing to coma has been reported (440). The condition is associated with dystonia, chorea, rigidity, and, at times, opisthotonus and external ophthalmoplegia. It is completely reversible by the administration of biotin (5 to 10 mg/kg per day). MRI shows increased signal on T2-weighted images within the central part of the caudate and in parts or all of the putamen. Its cause is unknown, and all enzyme assay results have been normal.
Some of the other, even less commonly encountered organic acidurias are summarized in Table 1.15, together with any distinguishing clinical features. In most conditions, however, symptoms are indistinguishable; they consist mainly of episodic vomiting, lethargy, convulsions, and coma. Laboratory features include acidosis, hypoglycemia, hyperammonemia, and hyperglycinemia. The organic acidurias can be diagnosed during an acute episode by subjecting serum or, preferably, urine to organic acid chromatography. In addition, a marked reduction in plasma-free carnitine exists, accompanied by an increased ratio of esterified carnitine to free carnitine. These assays should be performed on any child with neurologic symptoms who has an associated metabolic acidosis or who is
noted to have hyperglycinemia or hyperammonemia on routine biochemical analyses.
noted to have hyperglycinemia or hyperammonemia on routine biochemical analyses.
TABLE 1.15 Some of the Rarer Organic Acidurias of Infancy and Childhood | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


