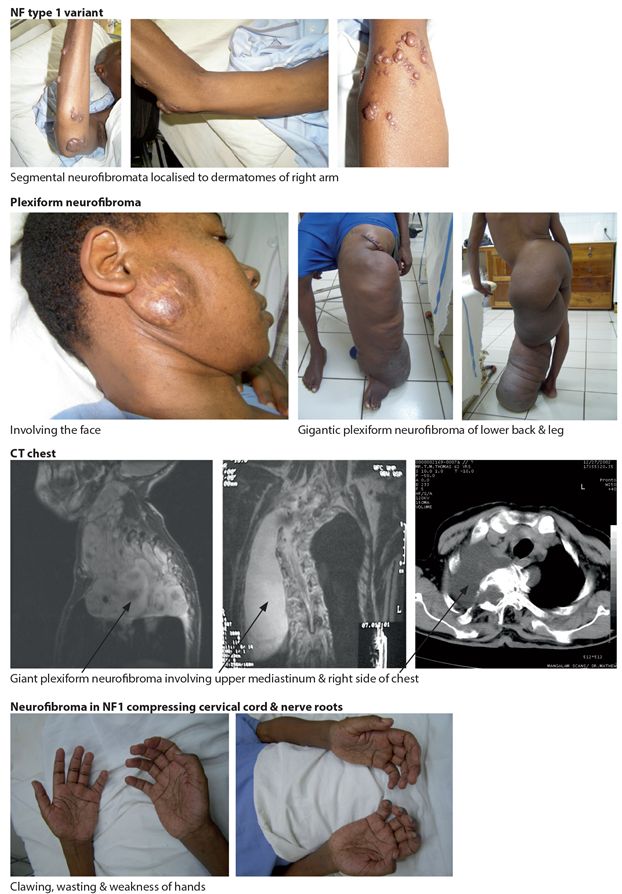
Figure 18.1 Clinical features NF type 1
Table 18.2 Diagnostic criteria for NF1
| Criteria: a diagnosis of NF type 1 is met if any two or more of the following is met. |
| 6 or more CAL* spots 1.5 cm or larger in post pubertal patients 0.5 cm or larger in prepubertal patients |
| 2 or more neurofibromata of any type or 1 or more plexiform neurofibroma |
| freckling in axilla, neck or groin |
| optic glioma |
| 2 or more Lisch nodules |
| a distinctive bone lesion dysplasia of the sphenoid bone dysplasia or thinning of long bone cortex (tibia) |
| first degree relative, independently diagnosed with NF1 |
* Café-au-lait
NEUROFIBROMATOSIS TYPE 2
Neurofibromatosis type 2 is a much rarer autosomal dominant disorder affecting about 1 in 50,000 due to a defect in the NF2 gene (merlin) on chromosome 22. It is characterised by few skin manifestations and the development of intracranial tumours, acoustic neuromas (vestibular Schwannoma) (Fig. 18.2) and meningiomas; see diagnostic criteria (Table 18.3). Deafness due to an acoustic neuroma involving the eighth cranial nerve is the main clinical presentation of type 2. This is usually accompanied by tinnitus, vertigo, ataxia, facial numbness and weakness. The clinical features at presentation are usually unilateral although the acoustic neuromas are commonly bilateral on neuroimaging. The diagnosis is confirmed by neuroimaging of the brain, usually MRI (Fig. 18.2). Management is either conservative by observation only or with deep X-ray therapy (DXT), or surgery for the tumours where possible. Genetic counselling is a very important part of management.
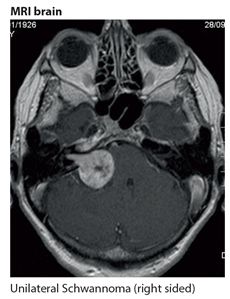
Figure 18.2 NF type 2
Table 18.3 Diagnostic criteria for NF2
| Criteria: a diagnosis of NF2 is made if one of the following is met. |
| bilateral vestibular Schwannoma, (either histologically or by MRI with contrast) |
| a parent, sibling or child with NF2 and either |
| a unilateral vestibular Schwannoma or 2 or more of meningioma, glioma, Schwannoma, posterior sub capsular lenticular opacities, cerebral calcifications |
| multiple meningiomas (2 or more) and one or more of glioma, Schwannoma, posterior sub capsular lenticular opacities, cerebral calcifications |
TUBEROUS SCLEROSIS
This is an autosomal dominant condition, due to mutations in either TSC1 on chromosome 9q (hamartin) or TSC2 on chromosome 16p (tuberin) with a prevalence of 1/15,000. Its frequency is not known in Africa but is most probably the same. About 60% of cases are due to new mutations. The main clinical features are the characteristic facial angiofibromata or adenoma sebaceum lesions on the cheeks and nose (Fig. 18.3), in combination with a history of epilepsy. There may be associated cognitive impairment. Additional skin manifestations include ash leaf hypopigmented macular type patches and the shagreen patch, a 1-10cm patch of orange peel like sub epidermal fibrosis found most often over the lumbar sacral area and subungual fibromas. CT shows a characteristic pattern of paraventricular subependymal calcified nodules or tuberi. Complications include tumours in the brain, hamartoma (Fig. 18.3) and astrocytoma. There are frequently other affected family members, most commonly siblings. Epilepsy may be difficult to control.
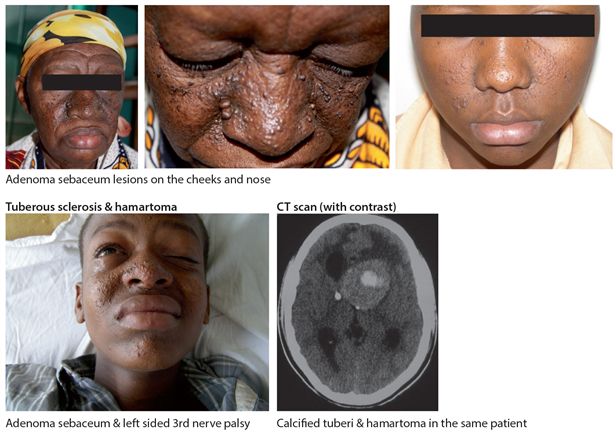
Figure 18.3 Tuberous sclerosis
STURGE WEBER SYNDROME
This is an uncommon sporadic disorder affecting 1/50,000 in high income countries. It is a neurocutaneous syndrome characterized by a port wine staining affecting one half of the face (Fig. 18.4) usually in a V1 distribution and associated neurological abnormalities. The defining characteristic of the syndrome is an underlying intracranial vascular abnormality in the leptomeninges affecting the cortical regions of the hemispheres. The syndrome includes seizures, glaucoma, headaches, behavioural problems and stroke like episodes. It has its onset usually in early childhood and is usually progressive. However the clinical course is highly variable and it is important to note that not all children or adults with the characteristic port wine facial staining have the syndrome. Neuroimaging, usually CT confirms the presence of intracerebral calcifications affecting the parietal or occipital lobes on the affected side but they can be generalised. Management is largely symptomatic and supportive.
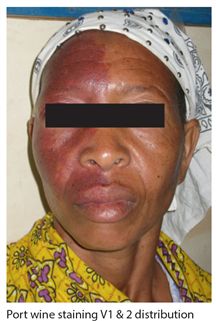
Figure 18.4 Sturge Weber syndrome
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


