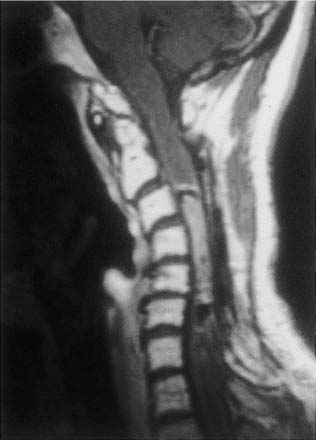
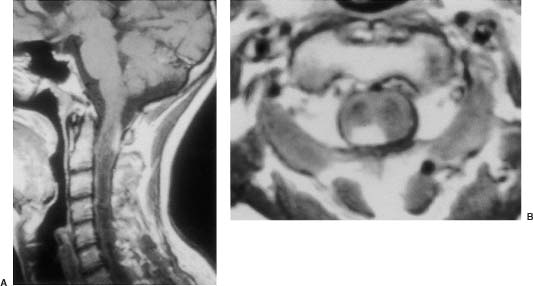
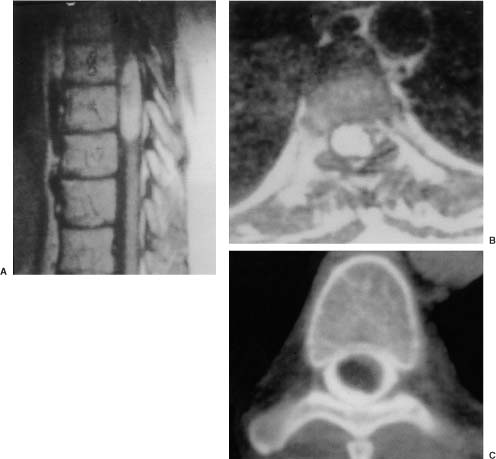
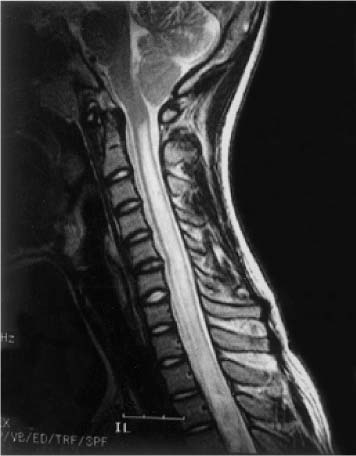
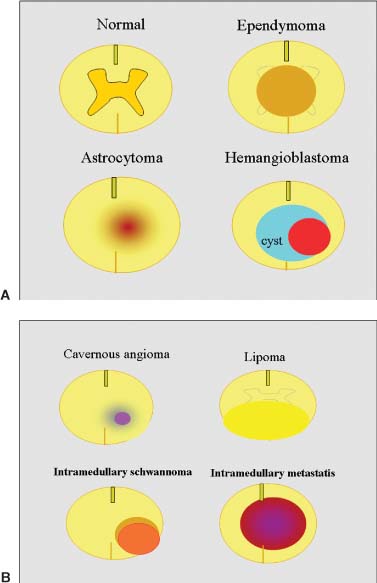
20 In one of the earliest attempts to excise an intramedullary spinal cord tumor, in 1911, Elsberg inadvertently made several nicks in the posterior aspect of the spinal cord while opening the dura. He noticed that the tumor began to extrude from these incisions. He connected the slits but made no attempt to excise the tumor. When he reopened the incision 1 week later, a plane had developed between the tumor and the surrounding spinal cord and the tumor was removed without difficulty.1 He subsequently published his experience, advocating this two-stage approach.2 Cushing3 also reported early success and excellent neurologic outcome with the resection of intramedullary spinal cord ependymomas. In spite of these early reports, intramedullary spinal cord tumor surgery in the first half of the 20th century produced dismal outcomes. As recently as 1969, Schneider4 asserted that when an intramedullary tumor is encountered that is not obviously cystic in a patient with little or no neurologic deficit, “the dura is left open with no attempt made to perform a myelotomy or procure a biopsy.” However, technical advances have resulted in decreased morbidity and mortality and improved neurologic outcomes, promoting renewed interest in aggressive management.5–7 The operating microscope is the most important of these, but bipolar cautery enhanced the ability to achieve a nearly complete tumor resection without inflicting injury to adjoining neural tissue, as Greenwood7 emphasized in his 1954 report of the total removal of six intramedullary spinal cord ependymomas. The ultrasonic tissue aspirator (CUSA(r); Valleylah, Boulder, Colorada) allows for debulking of firm tumors from the inside out, aiding in the development of a plane between tumor and spinal cord and minimizing manipulation of a spinal cord distended by tumor.8 The ultrasound and laser are also valuable surgical adjuncts.9 Magnetic resonance imaging (MRI) has revolutionized the diagnosis of intramedullary spinal cord tumors (IMSCT), allowing for earlier detection, better anatomic understanding of lesions, and accurate patient followup. Intramedullary tumors of the spinal cord are relatively rare and account for only 2 to 8.5% of all central nervous system tumors and 15% of primary intradural spinal tumors in adults.10–13 In the pediatric population, the proportion of intraspinal tumors that are intramedullary increases to at least 35%.14 IMSCT occur in all age groups but are uncommon in persons over 60 years of age 5.14,15 The mean age of patients with intramedullary tumors in one series was 38 years, with patients nearly equally divided between males and females.12 Tumors of glial origin predominate, comprising 68% of 1117 cases compiled by Fischer, Brotchi and coworkers. 16 In adults ependymomas are the most common intramedullary tumors, followed by astrocytomas and hemangioblastomas.11,12,17,18 In children astrocytomas are the most common intramedullary tumors, followed by gangliogliomas and mixed gliomas.10,19 Intramedullary spinal cord tumors may be found over the entire length of the spinal cord from the cervicomedullary junction to the conus medullaris. Astrocytomas in pediatric patients may involve the entire spinal cord.8,20 Patients with von Recklinghausen’s neurofibromatosis are predisposed to the development of IMSCT and may have intramedullary astrocytomas or ependymomas in addition to intradural extramedullary nerve sheath tumors; however, IMSCT are still uncommon in patients with neurofibromatosis type 1, occurring in only three of 1400 patients in one series.21 Similarly, patients with von Hippel-Lindau syndrome are predisposed to having multiple intramedullary hemangioblastomas. Spinal ependymomas arise from the ependymal lining of the spinal cord central canal or the cells of the ventriculus terminalis of the filum terminale.22 Ependymomas are most common in the cervical region.16,23 They grow slowly with an average interval of 16 months between onset of symptoms and diagnosis, although accelerated deterioration may be seen when tumor compression reaches a critical degree or from intratumoral hemorrhage. Pain (radicular or regional back pain) at the level of the tumor is the most frequent presenting symptom (65%). At presentation, an objective neurologic deficit is mild, most commonly consisting of sensory loss and weakness. Sensory loss may be “suspended” and identical to that seen with syringomyelia, or it may be asymmetrical. Atrophy of upper extremity musculature, particularly the hand intrinsic muscles, may be evident in cervical lesions.23 Bowel or bladder dysfunction is rare except in myxopapillary tumors of the conus, where it is reported in 20 to 25% of cases.24 Astrocytomas are a heterogeneous group of infiltrating tumors arising from neoplastically transformed astrocytes in both the brain and spinal cord. They are categorized in an ascending grading scale based on histopathologic evidence of anaplasia. They are histologically identical to astrocytomas occurring in the brain. Astrocytomas are the most common pediatric IMSCT, representing 59% of the tumors in a compilation of 13 series.25 In adults, they are second to ependymomas in frequency, accounting for ~20% of tumors.16,26 In distinction from intracranial astrocytomas, spinal cord astrocytomas tend overwhelmingly to be low-grade lesions in both children and adults. High-grade lesions (Kernohan grades III and IV) comprise only 10 to 15% of pediatric tumors and a slightly higher proportion in adults.27,28 There is a slight male predominance.16,29 The cervical area is most frequently affected, followed closely by the thoracic, although these lesions span an average of six spinal levels and may involve both regions.25 Rarely, the entire spinal cord may be involved.8 Presenting symptoms are variable and evolve over a period of months to years. Regional back pain and sensory disturbances are the most frequent complaints, with motor deficits evolving later.25,30 Because the motor representation in the spinal cord places hand fibers medial and leg lateral, a centrally located IMSCT may produce weakness in the hands before the lower extremity is affected. In young children, pain remains the most common symptom, but gait deterioration, motor regression, torticollis, and kyphoscoliosis are frequent presenting findings.15 In malignant tumors, pain is followed by a rapid evolution of symptoms, resulting in progression to significant disability in 3 to 5 months.27,31 Hemangioblastomas consist of thin-walled blood vessels interspersed with large, pale stromal cells and represent 3 to 11% of IMSCT. Up to one third of the cases occur in association with von Hippel-Lindau disease, and in such instances, multiple lesions may be present, particularly in the posterior fossa. The onset of symptoms is typically in the fourth decade of life, and the mean age at surgery is 40 years; childhood presentation is rare.30 There appears to be slight male predominance.32 The most frequent locations are thoracic (55%) and cervical (40%). Cyst formation occurs in 87% of cases.30 Sensory deficit, particularly loss of proprioception, is often the presenting symptom, as these lesions are generally found on the dorsal or dersolateral surface of the spinal cord. Pain or motor deficit is also frequently reported. There is a lengthy delay from the emergence of symptoms to surgery, reported as 4.2 years in one series.30 Intramedullary spinal cord lymphoma is an exceptional entity and is nearly always seen in association with acquired immunodeficiency syndrome (AIDS) or with lymphomas occurring in the brain. Symptoms tend to progress rapidly over a period of days to weeks. Lymphomas may enhance with gadolinium on MRI, but their appearance is otherwise not pathognomonic. There is increased cerebrospinal fluid (CSF) protein, but cytologic examination is nonspecific. If a diagnosis can be established by biopsy of coexisting brain lesions, surgery is contraindicated, as they are exquisitely sensitive to radiation and chemotherapy.33 Spinal lipomas extending intradurally frequently occur in patients with spinal dysraphism, but true intramedullary spinal cord lipomas are quite rare. Most patients present in the second to fourth decade of life, and there is no gender predilection. These tumors consist of ordinary adipose tissue. A long, indolent course ending with a rapid progression of symptoms is typical.34 Gangliogliomas are rare neoplasms, histologically distinct in containing both neoplastic neuronal and glial cells. They mainly occur in children and young adults, with both sexes affected in nearly equal proportion. They are slow-growing tumors that often produce gradual manifesting symptoms. Symptom and imaging characteristics fail to distinguish these lesions from other glial tumors.35–37 Cavernous angiomas or cavernomas represent 1 to 3% of IMSCT and are angiographically occult vascular malformations consisting of closely packed capillarylike vessels without intervening neural tissue. Half of all patients have multiple malformations. These lesions are not true neoplasms but produce symptoms as a result of hemorrhage. Patients with cavernomas present with two different clinical patterns. Slowly progressive myelopathy may result from repetitive, small hemorrhages with reactive gliosis or acute hemorrhage with rapid onset of neurologic impairment.38,39 Although it less uncommon for an individual hemorrhagic event to be catastrophic, once deficits appear the course is one of progressive deterioration, and surgery is the only effective treatment.40 Radiation therapy is ineffective in preventing recurrent hemorrhage. Patients with asymptomatic cavernomas found incidentally with MRI are not candidates for surgery; however, all symptomatic patients should be considered operative candidates, as progression of the neurologic deficit is the rule.39 Intramedullary spinal cord metastases are rare and account for 2 to 8% of all IMSCT.41,42 It is thought that the spinal cord is an infrequent site of metastases because of its small volume compared with the brain.43 A retrospective autopsy study of 627 patients with systemic cancer yielded 13 cases of intramedullary spinal cord metastasis. Four of these were instances of direct invasion of the spinal cord from leptomeningeal tumor, and the remainder were probably a consequence of hematogenous dissemination.44 The most common primary tumor sources are the lung and breast.45 In about one third of patients the spinal cord metastasis is the first manifestation of the presence of a primary tumor.46 Intramedullary spinal cord metastases present with rapidly progressive neurologic signs and symptoms; complete neurologic deficit occurs as early as 1 month after presentation in untreated patients. Patients tend to be older and frequently have extensive systemic disease. Median survival from diagnosis is 3 months.45 Ideal candidates for resection are patients with limited systemic disease and good neurologic function; however, patients with profound neurologic deficit or extensive systemic disease are unlikely to benefit from surgery. Radiation and corticosteroid therapy alone may be appropriate for lesions that are highly radiosensitive.47 Other rare tumors include dermoids, teratomas, oligodendrogliomas, and ganglioglioneurocytomas. Rosai-Dorfman syndrome is a rare extranodal manifestation of histiocytosis reported to present as an intramedullary spinal cord tumor.48,49 True intramedullary meningiomas and schwannomas are unusual, but case reports appear in the literature.30,50 Plain X-rays are normal in the majority of patients with IMSCT but may reveal an enlarged spinal canal with scalloping of the vertebral bodies, medial pedicle erosion, and thinning of the laminae.51 Scoliosis is common in children with IMSCT, often with the apex of the curvature to the left rather than the right, as is more commonly seen in idiopathic scoliosis. Magnetic resonance imaging is the preferred imaging modality for diagnosis of IMSCT and is generally the only study necessary. Images are first obtained in the sagittal plane followed by axial views through suspicious areas. IMSCT and associated cysts typically cause widening of the spinal cord. Tumors may be difficult to define from adjacent normal spinal cord, particularly on T1-weighted images. Tumor-associated cysts are usually hyperintense on T2-weighted images due to high protein content; however, most glial tumors and all hemangioblastomas enhance after administration of gadolinium. Prior to the advent of MRI, computed tomography (CT) myelography was the primary imaging modality for delineation of IMSCT. It could demonstrate spinal cord widening, but it could not be used to distinguish among an IMSCT tumor-associated cysts, and syringomyelia. Delayed CT scanning was sometimes used to demonstrate uptake of water-soluble contrast within the center of the spinal cord typical of tumor-associated cysts and syringomyelia. Spinal angiography may be considered when MRI suggests a hemangioblastoma. Although angiography delineates the location of the vessels that supply and drain the hemangioblastoma, the vascular supply is generally evident at surgery, and we have not found angiography to be important in planning or executing surgery. Ependymomas are characteristically isointense on T1-weighted images, hyperintense on T2-weighted images, and enhance strongly with administration of intravenous contrast (Fig. 20-1).50,52,53 Mixed signal intensity may be seen in areas of cyst or prior hemorrhage. The tumors characteristically occupy the center of the spinal cord. A hyperintense cap of hemosiderin on T1-weighted images is pathognomonic of ependymoma and is probably due to prior hemorrhage (Fig. 20-2). FIGURE 20-1 (A) Sagittal T1-weighted magnetic resonance imaging (MRI) of the cervical spine of a patient with a C4 ependymoma. The spinal cord is widened but the lesion is isointense in relation to the spinal cord. (B) After the administration of gadolinium-DTPA the T1 sagittal image demonstrates intense enhancement of the ependymoma at C4. (C) Axial T1 image at the C4 level after the administration of gadolinium-DTPA shows the lesion to be well defined and in the center of the spinal cord. FIGURE 20-2 Sagittal T1-weighted MRI of the cervical spinal cord after the administration of gadolinium-DTPA shows an enhancing lesion extending from C2 to C5. The hemosiderin cap at the superior margin of the tumor is characteristic of intramedullary ependymomas. Astrocytomas are iso- to slightly hypointense on T1-weighted images, hyperintense on T2-weighted images, and usually enhance after the administration of gadolinium, despite a low histologic grade.51 The degree of contrast enhancement is usually less than is seen with ependymomas, and astrocytomas are less well defined from surrounding normal spinal cord. Their location within the spinal cord is variable and may be central or eccentric. Tumor-associated cysts are common (Fig. 20-3). Although astrocytomas and ependymomas each have characteristic imaging findings, it is frequently not possible to definitively distinguish these two tumors from each other, and biopsy is the only way to make a definitive diagnosis.53 The imaging appearance of gangliogliomas on MRI is similar to that of astrocytomas; however, edema is characteristically absent, and enhancement is patchy and on the cord surface. The patients are typically younger, the tumors tend to be extensive in length, and tumor cysts are common. Bone erosion and scoliosis are frequently present, suggesting that these tumors are slowly growing. Hemangioblastomas demonstrate an intensely enhancing nodule after the administration of gadolinium. Tumor-associated cysts are frequently larger than the tumor itself and do not enhance. The cysts may extend several spinal levels and contain proteinaceous fluid, which is hyperintense on T2-weighted images. The lesions are usually located on the posterior surface of the spinal cord (Fig. 20-4). In patients with von Hippel-Lindau syndrome, multiple lesions are common, and the posterior cranial fossa and entire spinal cord should be imaged in a search for additional lesions. FIGURE 20-3 (A) T2-weighted image of an astrocytoma that extends from C4-C5 to T1. The spinal cord is swollen, and increased signal is patchy. (B) Axial T2-weighted image shows increased signal in an eccentric and peripheral fashion unlike the central location of ependymomas. FIGURE 20-4 (A) Sagittal T1-weighted MRI of the cervical spine following the administration of gadolinium-DTPA shows a small enhancing hemangioblastoma located dorsally at C2 with a large cyst involving the entire cervical spinal cord. (B) Corresponding axial image at C2 shows the dorsally located hemangioblastoma. Lipomas are hyperintense on T1, hypointense on T2, and do not enhance, and signal characteristics are identical to those of fat34 (Fig. 20-5). Cavernous angiomas contain hemorrhagic areas of various ages, and flecks of calcium may be seen on CT scans. The lesions do not enhance after the administration of gadolinium. MRI reveals a low-intensity rim of hemosiderin on T2 images with a heterogeneous core. The lesion itself is hyperintense on both T1 and T2 images. 38 Multiple central nervous system (CNS) lesions are frequently detected, particularly in familial cases. Although multiple sclerosis (MS) is not a neoplastic lesion, the unwary may mistake its imaging appearance on MRI for a neoplastic processes. MS plaques appear in the white matter involving one quadrant of the spinal cord such as the lateral, posterior, or anterior funiculi, in contrast to IMSCT, which are more centrally located, iso- to hypodense on T1-weighted scans, and hyperintense on T2.54,55 Spinal cord widening is minimal or absent. MS lesions are almost always limited to one or two spinal segments, in contrast to IMSCT, which typically span multiple levels (Fig. 20-6). A second noncontiguous lesion is unusual with spinal cord tumors but more common with demyelinating disease. IMSCT characteristically widens the spinal cord considerably, whereas the widening seen with MS is trivial or absent. Cysts are never seen in association with MS plaques.56,57 Lesions enhance with gadolinium during periods of active demyelination. If acute MS is suspected, a repeat scan 4 to 6 weeks after the initial study will show decreased mass effect, diminished area of hyperintensity on T2-weighted images, and diminution or disappearance of enhancement. Magnetic resonance imaging of the brain should be obtained in any patient suspected of having spinal cord multiple sclerosis. The presence of additional cranial lesions strongly supports the diagnosis of MS.53,58 The CSF usually tests positive for oligoclonal bands, and visual evoked responses are abnormal in patients with MS. FIGURE 20-5 (A) Sagittal T1-weighted MRI of the thoracic spine shows hyperintense signal characteristic of an intramedullary lipoma. (B) Corresponding axial image confirms the intramedullary location of the lesion. (C) Axial CT scan of the same patient shows the hypodense lipoma displacing the spinal cord to the right and anteriorly. Multiple sclerosis in its acute stage can present with a clinical picture and lesions on imaging studies that resemble IMSCT. A careful history, however, frequently reveals the correct diagnosis. Although intratumoral hemorrhage, in rare instances, can produce rapid neurologic deterioration followed by partial recovery in patients with IMSCT, the more typical course is gradual and progressive neurologic deficit. MS, however, presents with rapid onset of neurologic dysfunction followed by partial or total recovery over days to weeks. The imaging findings of MS described in a previous section are dissimilar enough from IMSCT that there is generally little confusion between the two entities Syringomyelia, a cystic cavitation that widens the spinal cord, occurs in congenital conditions like the Chiari malformation or after trauma. It presents clinically with symptoms of pain, sensory loss (particularly pain and temperature modalities), muscular atrophy, weakness, and myelopathy. The clinical course is more insidious than that of IMSCT, but in all other respects the manifestations of the two diseases may be indistinguishable. MRI, however, usually provides the correct diagnosis. The fluid in the syrinx has a signal similar to that of CSF. After the administration of gadolinium the walls of a syrinx do not enhance, whereas cystic intramedullary tumors such as hemangioblastomas or ependymomas demonstrate enhancement. In patients with syrinxes occurring in association with the Chiari malformation visualization of the cervicomedullary junction with sagittal MRI reveals tonsillar herniation (Fig. 20-7); however, syrinxes may also occur without tonsillar herniation in patients who have arachnoiditis or who have sustained spinal cord trauma. FIGURE 20-6 (A) Sagittal T1-weighted MRI of the cervical spinal cord following the administration of gadolinium-DTPA in a patient with acute multiple sclerosis showing slight enhancement at the C2-C3 level. There is virtually no widening of the spinal cord. (B) Axial T1-weighted MRI following the administration of gadolinium-DTPA shows characteristic quadrantic location of enhancement in the dorsolateral region of the cervical spinal cord. Sarcoidosis is a multisystem disorder of unknown origin with noncaseating granulomatous lesions that affects adults between the ages of 20 and 40 years. Patients with pulmonary sarcoidosis frequently present with bilateral hilar adenopathy and pulmonary infiltrates. Central nervous system involvement by sarcoidosis, a systemic disease, occurs in 5 to 10% of cases, and on rare occasions intramedullary spinal cord involvement is the first manifestation of the disease. Presenting symptoms may be regional pain and sensory or motor disturbance. T2-weighted MRI reveals an extensive high-intensity area in the spinal cord, and gadolinium-diethylenetriamine pentaacetic acid (Gd-DTPA) MRI shows a patchy multifocal enhancement of the spinal cord (Fig. 20-8). There is sometimes linear peripheral enhancement along the anterior median fissure or posteromedian sulci59 thought to be due to inflammatory changes in the leptomeninges. Laboratory abnormalities include hyperglobulinemia, an elevated serum angiotensinconverting enzyme level, and evidence of depressed cellular immunity manifested by cutaneous anergy. Although CSF levels of angiotensin-converting enzyme are frequently elevated in sarcoidosis of the brain, levels were normal in the reported cases of intramedullary spinal cord lesions. Definitive diagnosis is made only with a surgical biopsy. Treatment is a prolonged course of corticosteroids.60 FIGURE 20-7 T2-weighted sagittal MRI showing extensive hyperintense signal within the spinal cord in a patient with a syrinx. The herniation of the cerebellar tonsils through the foramen magnum is characteristic of the Chiari malformation and is helpful in distinguishing this entity from a tumor-associated cyst. The objectives of surgical intervention are to provide total tumor removal while preserving or improving neurologic function, and to obtain a tissue diagnosis. The infiltrating nature of some lesions, however, may make total removal impossible without an unacceptable loss of neurologic function. In such instances, subtotal resection may still be a worthwhile goal to reduce tumor mass in preparation for adjunctive therapy. In young children, consideration should be given to radical resection to defer radiation treatment with its deleterious effects on the developing nervous system. FIGURE 20-8. T1-weighted Gd-DTPA enhanced sagittal MRI demonstrates patchy multifocal enhancements in the spinal cord characteristic of sarcoidosis. The natural history of IMSCT is that of progressive neurologic deficit, and early operative intervention is desirable, as the postoperative functional outcome is closely correlated with the severity of the patient’s preoperative deficit.15,61 Ideal surgical candidates are those who are ambulatory or have minimal neurologic deficit. Even patients with significant deficit may still derive benefits from surgery with preservation of sphincter function or the ability to position in bed. Patients with complete neurologic loss of function are not appropriate surgical candidates. Patients who are potential surgical candidates must be counseled to maintain realistic expectations of surgical outcomes, including worsening of residual neurologic function. In cases where there are very slowly progressive and minor motor or sensory deficit, both the surgeon and the patient are frequently and understandably reluctant to consider an operation. This is especially so if imaging studies suggest the presence of an infiltrating tumor, such as an astrocytoma, which usually cannot be totally removed and may frequently result in permanent increase of neurologic deficit. When imaging studies suggest the presence of an ependymoma, the patient can be advised that cure is likely. Because the tumors are generally not infiltrative, a permanent increase in neurologic deficit is infrequent. Incidentally discovered cavernous malformations should be followed, as the natural history of asymptomatic lesions is not completely understood. If a patient has acutely deteriorated secondary to hemorrhage, it is better to wait several weeks before surgery to permit recovery. 40 Imaging studies are analyzed to precisely delineate the tumor’s solid and cystic components and to distinguish them from spinal cord edema. The levels and extent of the anticipated laminectomy are noted. We place patients on 64 mg of methylprednisolone every 6 hours for 24 hours prior to surgery. Methylprednisolone administration is continued in the postoperative period at the same dose for 48 hours and gradually tapered over the next 5 days. If lower extremity motor deficits exist, Doppler studies of the deep lower extremity venous system are performed. If deep venous thrombosis is identified, a vena caval filter is placed. If the deep venous system is normal, antiembolism compression stockings are placed immediately prior to operation and continued in the postoperative period until the patient is ambulatory. Most surgeons employ evoked potential monitoring in the hope that intraoperative data will guide the extent of the surgical resection and predict postoperative deficits. Somatosensory-evoked potentials (SSEPs) monitor the integrity of the dorsal columns. Significant intraoperative changes in SSEPs are demonstrated to be predictive of postoperative neurologic deficits.62 Their usefulness in improving outcome, however, may be limited.63 Furthermore, preoperative neurologic deficit may result in failure to obtain baseline readings. Variability in user skill and equipment error can be other causes of unsatisfactory physiologic data. Most importantly, however, neurologic damage can result in the brief, 10- to 60-second delay between the time of spinal cord injury and evoked potential changes generated from computer averaging techniques. Such neurologic injury may be irreversible, in distinction from evoked potential changes seen in the course of scoliosis surgery, which are usually reversible by repositioning the instrumentation. Motor-evoked potentials (MEPs) are a newer technique used to assess the integrity of the corticospinal tracts during IMSCT surgery and provide “real-time” intraoperative data. Utilizing scalp electrodes in combination with epidural electrodes, the presence of MEPs is believed to correlate better with surgical outcome than the preoperative neurologic exam.64 One surgical group reports the use of a 50% decline in the amplitude f MEPs as an indication to interrupt dissection.65 The benefit of this technique, however, was limited by the fact that MEPs could not be measured in a large proportion of patients, many of whom were those who had baseline neurologic compromise and stood to benefit most from such monitoring.66 Kothbauer et al66 believe that evoked potential monitoring is an essential adjunct to surgery. The use of evoked potential monitoring facilitates the prediction of outcome after surgery.64 However, controlled case studies support the efficacy of evoked potential monitoring in improving the outcome from surgery are lacking. In short, although we employ both MEP and SSEP monitoring, we are not convinced by our own experience or by the data from the literature that their use results in improved outcome. Patients are placed prone upon gel rolls on the operating room table. For cervical or high thoracic lesions, the head is placed in three-point fixation, with the neck in a neutral position. For lesions at or below T3, the head may be turned to the side and rested on a cushion, taking particular care to protect the eyes from pressure or abrasions. The horseshoe headholder and the knee-chest position are avoided due to the respective risks of skin breakdown and rhabdomyolysis.67 Laminectomy is limited to one vertebral level above and below the tumor; ultrasound confirms the extent of solid tumor and bone removal is extended as necessary.9 Intramedullary tumors are characteristically hyperechoic in their solid portion and hypoechoic in their cystic/ necrotic regions.68 It is unnecessary to expose rostral or caudal cystic components of the lesion, as cyst fluid is produced by tumor cells rather than the nonneoplastic cyst wall; tumorassociated cysts resolve after complete tumor removal.5 Laminoplasty (osteoplastic laminotomy) is utilized by pediatric neurosurgeons to prevent the development of postoperative spinal deformity.14,69,70 Adult patients are at low risk of postoperative spinal deformity, and laminoplasty is not performed. Posterior instrumentation of the cervical spine is used only if kyphotic deformity is present preoperatively.
Intramedullary Spinal Cord Tumors
 History
History
 Epidemiology and Presentation
Epidemiology and Presentation
 Presentation
Presentation
Ependymomas
Astrocytomas
Hemangioblastomas
Lymphomas
Lipomas
Gangliogliomas
Cavernous Angiomas
Metastases
Other Tumors
 Diagnostic Imaging Studies
Diagnostic Imaging Studies
Plain X-Rays
Magnetic Resonance Imaging
Computed Tomography/Myelography
Spinal Angiography
Imaging Characteristics of Specific Diagnostic Entities
Ependymomas
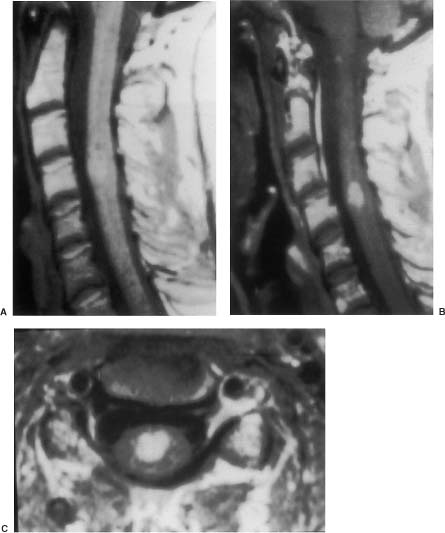
Astrocytomas
Gangliogliomas
Hemangioblastomas
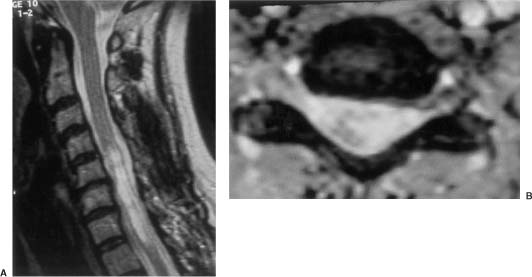
Lipomas
Cavernous Angiomas
Multiple Sclerosis
Differential Diagnosis
Multiple Sclerosis
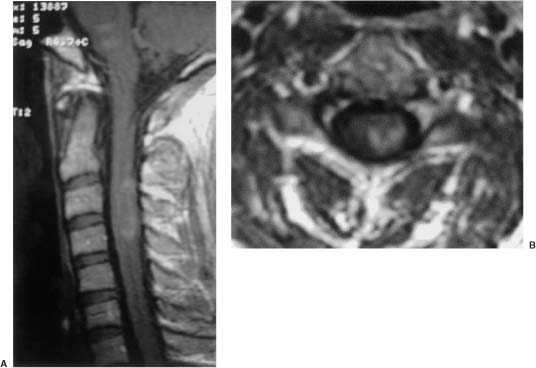
Sarcoidosis
 Surgical Management
Surgical Management
Goals
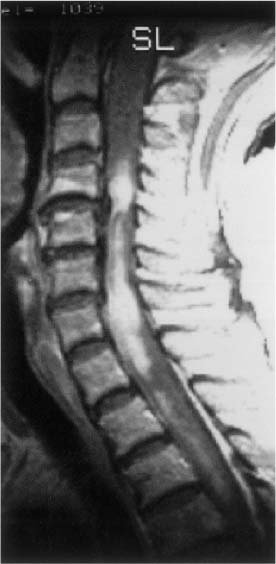
Selection of Operative Candidates
Perioperative Management
Evoked Potential Monitoring
Patient Positioning
Operative Technique
General Considerations
Related posts:
 Sacral Tumors: Primary and Metastatic
Sacral Tumors: Primary and Metastatic
 Spinal Stereotactic Radiosurgery
Spinal Stereotactic Radiosurgery
 Anatomy of the Spine and Spinal Cord
Anatomy of the Spine and Spinal Cord
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


