Lesionectomy: Management of Substrate-directed Epilepsies
Kurupath Radhakrishnan
Itzhak Fried
Gregory D. Cascino
Introduction
Nearly one third of patients with newly diagnosed epilepsy on long-term follow-up will have their seizures unsatisfactorily controlled by treatment with antiepileptic drugs.125 A majority of these patients with difficult-to-control seizures have focal epilepsies.20 Nearly half of the patients with medically refractory focal epilepsies are potential candidates for epilepsy surgery.44 The remarkable advances in neuroimaging technologies during the past two or three decades has allowed detection of a variety of brain lesions responsible for refractory focal epilepsies such as hippocampal sclerosis, malformations of cortical development, neoplasms, vascular malformations, and focal gliotic lesions that are amenable to surgical treatment. Table 1 summarizes the frequently encountered surgically remediable lesional epilepsy syndromes. The understanding that a majority of patients with substrate-directed intracranial lesion–associated chronic focal epilepsies can be selected for surgery based on a noninvasive presurgical evaluation has resulted in recent years in both an increase in the number of epilepsy surgeries being performed in developed countries and in the creation of epilepsy surgery programs in developing countries. A recent survey revealed that in 26 of 142 (18.3%) economically disadvantaged nations, at least one center regularly conducted epilepsy surgery, compared to 18 of 24 (75%) developed countries.149
In this chapter, we willc review the etiologies and pathogenesis of refractory lesional epilepsies and discuss the surgically remediable lesional syndromes including the presurgical evaluation strategies, surgical management, and postsurgery outcomes. The syndrome of mesial temporal lobe epilepsy with hippocampal sclerosis, which is the prototype of medically refractory lesional human epilepsy syndromes, is discussed in Chapter 247. Therefore, we will confine our discussion to the remaining lesional epilepsy syndromes listed in Table 1.
Historical Perspectives
Lesionectomy was introduced as one of the earliest surgical treatments for patients with partial or localization-related epilepsy. The modern era of epilepsy surgery began on May 25, 1886, when Victor Horsley operated, at the National Hospital for Paralysed and Epileptic at Queen Square in London, on a 22-year-old male referred to him by John Hughlings Jackson with focal motor seizures due to a focal cortical scar left behind by a depressed skull fracture he sustained 15 years before.65 A month later, on June 22, 1886, Horsley operated on a second patient of Jackson’s, a 22-year-old male who for 2 years suffered from recurrent focal motor seizures with onset in the left thumb and forefinger progressing on to involve the rest of the left upper extremity and left half of the body, the so-called jacksonian seizures.65 At exploratory operation, Horsley found a tumor at the site of brain predicted by Jackson based on the seizure semiology that turned out to be a tuberculoma. This second operation is truly the first substrate-directed surgical treatment of epilepsy, not only because it was guided by the meticulous analysis of the seizure semiology, but also because it involved resection of the lesion and the surrounding brain tissue responsible for the seizures.
The introduction of electroencephalography (EEG) by Hans Berger in the 1930s permitted identification of epileptiform discharges.12 Herbert Jasper, in collaboration with Wilder Penfield, effectively utilized EEG in the neurosurgical treatment of focal epilepsies at the Montreal Neurological Institute, not only in preoperative localization of the epileptogenic focus, but also during surgery to tailor the extent of resection—intraoperative electrocorticography.69 The development of inpatient video-EEG recordings and digitalization of the EEG data in the 1980s and 1990s permitted precise electroclinical correlation of focal seizures.22
Although the introduction of computed tomography (CT) in the 1970s facilitated detection of some overt lesions,53 the most important advance in the surgical treatment of focal epilepsies is the introduction of magnetic resonance imaging (MRI) in the 1980s.83 A high-resolution MRI has become the integral part of presurgical evaluation of focal epilepsies, as it is the only investigation that can distinguish between substrate-related and substrate-unrelated epilepsies. Several studies, both in temporal and extratemporal epilepsies, have shown that an MRI-identified lesion is a strong predictor of favorable seizure outcome following surgery.23,93 Newer MRI techniques like MR spectroscopy, MR volumetry, MR T2 relaxometry, and diffusion-weighted MRI have further improved the yield of detection of the epileptogenic focus.41 A wide array of additional investigations such as positron emission tomography (PET), single photon emission tomography (SPECT), functional MRI (fMRI), MR tractography, and magnetoencephalography (MEG) and the coregistration of data from multiple sources have helped not only in detecting areas of altered electrical activity, blood flow, and metabolism, but also in defining the relationship of the lesion to adjacent cortical areas and white matter tracts with critical neurologic function.43,87,113,136
Table 1 Surgically Remediable Lesional Epilepsy Syndromes | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
New surgical techniques such as microneurosurgery and image-guided surgery have made lesionectomy safer and more effective.30 The role of radiosurgical techniques in selected substrate-related focal epilepsies such as those due to vascular
malformations129 and hypothalamic hamartomas117 are currently undergoing evaluation.
malformations129 and hypothalamic hamartomas117 are currently undergoing evaluation.
Pathophysiology of Substrate-directed Epilepsies
The underlying pathophysiologic mechanisms of lesion-related epilepsies are multifactorial and poorly understood. While focal malformations of cortical development17,108 and hypothalamic hamartomas72 are intrinsically epileptogenic, in brain tumors and focal gliotic lesions, it is often the alterations in the perilesional cerebral cortex that incite epileptogenesis. Occasionally, long-standing lesions within or adjacent to the temporal lobe can result in hippocampal sclerosis. In these patients with dual pathology, either the primary lesion or the secondary hippocampal sclerosis or both may initiate epileptic seizures.25,27,46,49
Several histologic changes have been observed in the brain tissue immediately surrounding a brain tumor, but it remains unclear how these factors relate to epileptogenesis.127 Comparison of peritumoral cortex from patients with and without seizures have demonstrated significant differences in the number and organization of the synapses with an increase of excitatory synapses94 and a decrease of inhibitory synapses.59 Wolf et al.151 investigated perilesional changes in pharmacoresistent tumoral epilepsies by using the immunoreactivity of the γ-aminobutyric acid (GABA)A receptor, NR1-N-methyl-D-aspartate (NMDA) receptor subunits, and glutamate decarboxylase activity and concluded that not only alterations of concentrations of neurotransmitters in peritumoral cortex occur, but also that these changes involve heterogeneously the number of receptors and the concentration of enzymes that synthesize and degrade neurotransmitters. Additionally, the multidrug resistance (MDR) protein-1 and the MDR-associated proteins may play a role in contributing to pharmacoresistance.88,135 Recently, the CC genotype at the C3435 T polymorphism of the MDR-1 gene was shown to be associated with resistance to antiepileptic drugs.131
Glioneural tumors like ganglioglioma and dysembryoplastic neuroepithelia tumor are frequently associated with dysplasia in the adjacent cortex. In a recent study, in 20 of 24 (83%) surgically treated cases of dysembryoplastic neuroepithelial tumor (DNET), associated cortical dysplasia was found on pathologic examination.141 To achieve favorable postoperative seizure outcome, presurgical evaluation and surgical treatment in these lesions should be aimed at defining and radically resecting the pathologic areas, both the tumor and associated dysplasia. Similarly, in an analysis of the seizure outcome of a group of patients with refractory focal epilepsies associated with cavernous malformations, complete removal of surrounding hemosiderin-stained brain tissue along with the vascular lesion was shown to result in a better seizure outcome when compared to removal of the lesion alone.9
Substrate-directed Epilepsy Syndromes
Tumoral Epilepsies
Brain neoplasms comprise 10% to 30% of the pathologic substrate in surgically treated patients with medically refractory focal epilepsies.21,138,150 The histopathologic profile of tumor-related chronic epilepsy is quite different from the usual pattern of primary brain neoplasms. Although low-grade tumors such as ganglioglioma and DNET account for only a very small proportion of primary brain neoplasms, they are more frequently associated with tumoral refractory epilepsies.74,91,98,109
Ganglioglioma
The recent World Health Organization classification on brain tumors defines ganglioglioma as a neoplasm composed of neoplastic neural and neoplastic glial cells (Fig. 1).75 Ganglioglioma may occur in any part of the neuraxis, but the most frequent site is the temporal lobe. Ganglioglioma is a common cause of tumor-related refractory epilepsies and in some series account for nearly half of the tumoral substrates,54,91,98,109 though in others it is lower.18,50 In one series of 38 patients with ganglioglioma (28 temporal and 10 extratemporal), the mean age at surgery and mean duration of epilepsy prior to surgery was 19.3 years and 9.7 years, respectively.99 In another series of 23 patients with surgically treated ganglioglioma, the median age at surgery was 20 years and the median duration of epilepsy prior to surgery was 9 years.116
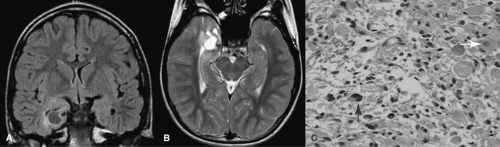 FIGURE 1. Imaging and histopathologic data of an 11-year-old boy with complex partial seizures since age 1½ years. A: Coronal fluid attenuated inversion recovery magnetic resonance imaging sequence shows a cystic space-occupying lesion involving the right hippocampus and parahippocampal gyrus with surrounding areas of hyperintensity. B: T2-weighted spin echo sequence shows extension of the lesion to the amygdala and head of hippocampus. C: Photomicrograph shows admixture of neoplastic neurons (white arrow) and astrocytes (black arrow) characteristic of ganglioglioma (hematoxylin and eosin ×200). The patient was seizure free for 18 months following anterior temporal lobectomy with amygdalohippocampectomy. |
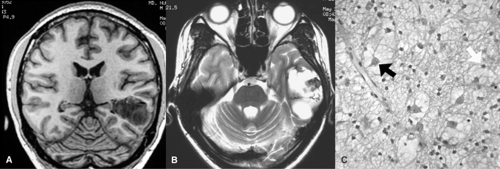 FIGURE 2. Imaging pathologic correlation in a 20-year-old male with complex partial seizures with behavioral arrest, oral and bilateral upper automatisms, head and eye deviation to the right, and frequent secondary generalization. A multicystic mass lesion involves the left inferior temporal gyrus with remodeling of the overlying skull, which is hypointense on T1 coronal sequence (A) and hyperintense on axial T2 sequence (B) with hypointensity within suggestive of calcification. C: Photomicrograph shows microcystic spaces with floating neurons (black arrow) and oligodendrocyte-like cells (white arrow), characteristic of a dysembryoplastic neuroepithelial tumor (hematoxylin and eosin ×200). Patient was seizure free for the last year following lesionectomy. |
The typical CT appearance of gangliogliomas is a well-circumscribed hypodense or isodense lesion, with focal calcification in one third of tumors and enhancement in nearly half.52 With MRI, they appear as a heterogenous mass, hypointense on T1-weighted and hyperintense on T2-weighted sequences (Fig. 1). Cystic changes and slight enhancement are frequently observed.52
Dysembryoplastic Neuroepithelial Tumor
The DNET accounts for 10% to 20% of chronic tumoral epilepsy syndromes.35,91,109 Its typical pathologic features include disorganized arrangement of neural and glial elements
without significant cytological atypia35 (Fig. 2). The frequent association of DNET with cortical dysplasia is being increasingly recognized.114,141 From these features, DNET might be regarded even as an atypical hamartomatous lesion.35 The histologic diagnosis of DNET may be challenging in the absence of a sizeable specimen preserving the peculiar disarray of the neural and glial elements. In a group of 39 patients with DNET, age at onset of seizures ranged from 1 to 19 years (mean 9 years), and the duration of epilepsy prior to surgery averaged 9 years (range 2 to 18 years).35 In a recent Japanese series of 20 patients, the mean age at seizure onset was 16.8 years (range 1 to 38 years) and the mean age at surgery was 24 years (range 4 to 46 years).141
without significant cytological atypia35 (Fig. 2). The frequent association of DNET with cortical dysplasia is being increasingly recognized.114,141 From these features, DNET might be regarded even as an atypical hamartomatous lesion.35 The histologic diagnosis of DNET may be challenging in the absence of a sizeable specimen preserving the peculiar disarray of the neural and glial elements. In a group of 39 patients with DNET, age at onset of seizures ranged from 1 to 19 years (mean 9 years), and the duration of epilepsy prior to surgery averaged 9 years (range 2 to 18 years).35 In a recent Japanese series of 20 patients, the mean age at seizure onset was 16.8 years (range 1 to 38 years) and the mean age at surgery was 24 years (range 4 to 46 years).141
The CT demonstrates a well-circumscribed hypodense lesion, but may be normal in 10% of cases.52 The MR appearance of DNET may be indistinguishable from that of ganglioglioma, astrocytoma, and oligodendroglioma. Absence of calcification, the cortical location and associated gyral thickening, and frequent cystic changes may be helpful in differentiation52 (Fig. 2).
Low-grade Glial Neoplasms
Pilocytic astrocytoma, low-grade gliomas, pleomorphic xanthoastrocytoma, and oligodendroglioma comprise a substantial part of the pathologic substrates of tumoral chronic epilepsies.18,50,91,109 Seizures occur in 70% of those with astrocytomas and in 92% of those with oligodendrogliomas.16,138 Nearly 75% of glial neoplastic lesions associated with chronic epilepsy involve the temporal lobe.16,98,138 Fried et al.50 have
proposed that limbic and neocortical gliomas associated with chronic seizures constitute a unique group of glial tumors that involve the gray matter, arise in a young host, and exhibit stable biologic behavior over many years. Yet the possibility of malignant transformation cannot be ruled out. This is an added consideration in the treatment of these lesions, as their total re-moval addresses the issues of both seizure and tumor control.
proposed that limbic and neocortical gliomas associated with chronic seizures constitute a unique group of glial tumors that involve the gray matter, arise in a young host, and exhibit stable biologic behavior over many years. Yet the possibility of malignant transformation cannot be ruled out. This is an added consideration in the treatment of these lesions, as their total re-moval addresses the issues of both seizure and tumor control.
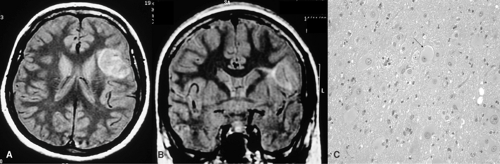 FIGURE 3. The data of a 19-year-old female with frequent complex partial seizures characterized by cephalic sensation, bilateral upper limb clonic jerks, and falls. A: Axial proton density (PD) magnetic resonance imaging sequence shows thickened left inferior frontal gyrus forming a globular hyperintense lesion. B: Coronal PD sequence depicts a radial band extending transmantally up to the frontal horn of left lateral ventricle. C: Photomicrograph shows dysplastic neurons with variation in size and shape and balloon cells (arrow). There were no postoperative neurologic deficits and no disabling seizures for the last 4 years following lesion resection under awake craniotomy and cortical stimulation and mapping. |
Malformations of Cortical Development
Malformations of cortical development are second only to hippocampal sclerosis as a frequent pathologic substrate detected in 20% to 40% of pediatric and adult surgically treated patients with pharmacoresistent epilepsies.115,152 Developmental malformations are fully discussed in Chapter 259. The main pathologic hallmark of malformations of cortical development is the presence of columnar and laminar disorganization that can be intermixed with various cellular abnormalities that include dysmorphic neurons, giant neurons, and balloon cells.115 The classification scheme of Barkovich et al.7 is centered on three processes of cerebral development: Neuronal proliferation and eventual apoptosis of selected cells; neuronal migration; and cortical organization. In relation to resective surgery, the following malformations of cortical development are relevant: Focal cortical dysplasia or more appropriately focal malformations of cortical development, periventricular heterotopia, polymicrogyria, and schizencephaly.100,134 In addition, malformations of cortical development associated with DNET and tuberous sclerosis significantly contribute to the pathogenesis of refractory focal epilepsies.
Focal Malformations of Cortical Development
The focal malformations of cortical development are important substrates for focal motor status epilepticus or epilepsia partialis continua. Preoperative detection of focal malformations of cortical development depends on high-resolution MRI. The following MRI features suggest focal malformations of cortical development: Local cortical thickening, blurring of gray–white matter interphase, increased signal in the underlying white matter, and sometimes a wedge-shaped tail extending radially to the ventricle (“transmantle dysplasia”) (Fig. 3).6 Distinction from tuberous sclerosis–associated lesions may not be possible and may not even be rationale.10,92 Focal malformations of cortical development are most frequently encountered in the frontal or temporal lobes, though may occur anywhere.
Periventricular Heterotopia
This is a form of focal malformations of cortical development in which neurons generated in the periventricular region have failed to migrate, resulting in nodules abutting the ventricular ependymal lining. Isointensity of periventricular nodules with normal gray matter on all sequences of MRI helps to distinguish periventricular nodular heterotopia from the subependymal nodules of tuberous sclerosis.5 The pathophysiologic basis of epilepsy in periventricular nodular heterotopia may be related to their intrinsic epileptogenicity and connection to other nodules and to the cortex.80
Polymicrogyria and Schizencephaly
Polymicrogyria is the presence of an excess number of abnormal small gyri resulting in an irregular cortical surface. Polymicrogyria is not invariably associated with epilepsy. Patients may present with developmental delay, learning disabilities, or congenital hemiparesis.134 The mechanism of epileptogenesis in polymicrogyria is unknown; surrounding cortex is implicated in epileptogenesis. Perhaps because of this reason, surgical resection of the microgyric area alone seldom results in favorable seizure outcome. Schizencephaly is currently grouped with polymicrogyria, which is defined by the presence of transcortical cleft, open or closed, lined by gray matter, and often with microgyria along the cleft borders.7 Schizencephaly may be associated with a wide range of other malformations involving the septum, optic nerve, and hippocampus.61
Tuberous Sclerosis Complex
Tuberous sclerosis complex is an autosomal dominantly inherited multisystem genetic disorder characterized by variable phenotypic expression, resulting from mutations in one of two genes, TSC1 on chromosome 9q34 encoding hamartin, and TSC2 on chromosome 16p13 encoding tuberin.33 The brain lesions in tuberous sclerosis complex are due to disordered
neurogenesis and neuronal migration, resulting in very different neurologic phenotypes including seizures, mental retardation, learning disabilities, and autism. Pathologically, tubers are characterized by disorganized cortical laminations, aberrant dendritic and axonal projections and arborizations, astrocytic proliferation, dysplastic neurons, and giant cells.32 Giant cells are the pathognomonic histologic feature of tuberous sclerosis complex.33
neurogenesis and neuronal migration, resulting in very different neurologic phenotypes including seizures, mental retardation, learning disabilities, and autism. Pathologically, tubers are characterized by disorganized cortical laminations, aberrant dendritic and axonal projections and arborizations, astrocytic proliferation, dysplastic neurons, and giant cells.32 Giant cells are the pathognomonic histologic feature of tuberous sclerosis complex.33
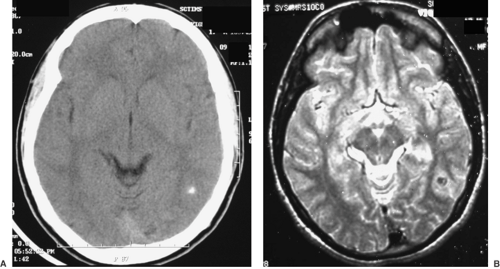 FIGURE 4. A 17-year-old male referred to the Comprehensive Epilepsy Program of the Sree Chitra Tirunal Institute for Medical Sciences and Technology, Trivandrum, India, from an endemic area for cysticercosis in southern India for refractory complex partial seizures since the age of 14 years. A: Noncontrast axial computed tomography shows a calcified lesion in the left posterior temporal region. B: T2 axial magnetic resonance imaging sequence shows hypointense calcification with surrounding hyperintensity representing gliosis. The patient was seizure free for nearly 2 years following lesionectomy. |
Epilepsy is the most common presenting symptom of tuberous sclerosis complex. The seizures are focal or multifocal in origin and are often resistant to antiepileptic drugs.105 Immunohistochemical evidence and electrophysiologic evidence have indicated that the neurons within cortical tubers are intrinsically epileptogenic.33 Surgical management of epilepsy is challenging in patients with tuberous sclerosis complex because the epileptogenic tubers are often multiple, bilateral, and extratemporal, overlapping with eloquent cortex.57,68,82
Hypothalamic Hamartomas
With improved detection of the lesion by MRI, the wide clinical spectrum of epilepsy syndromes associated with hypothalamic hamartoma is being increasingly understood. In addition to gelastic seizures, hypothalamic hamartoma can manifest with partial seizures of temporal or frontal semiology and with a catastrophic pediatric epileptic encephalopathy with polymorphic seizures, intellectual deterioration, and marked behavioral disturbances.8,72,139 The seizures in individuals with hypothalamic hamartoma are often refractory to antiepileptic drugs, but in most cases are reversed by surgical excision,48,103 radiofrequency thermocoagulation, or Gamma Knife surgery (Chapter 250).117
Vascular Malformations
Seizures occur in about one third of patients with arteriovenous malformations and the response to antiepileptic drug therapy is variable.64 Among those with medically refractory focal seizures, nearly three fourths of patients benefit either from lesion resection111 or from radiosurgery.129 Seizures represent the most common symptomatic presentation of supratentorial cerebral cavernous malformations.31,73 In a comparative analysis of the seizure propensity of patients harboring arteriovenous and cavernous malformations, Awad et al.4 found a prevalence of 50% to 70% in cavernomas and 20% to 40% in arteriovenous malformations. Recurrent microhemorrhages and the resultant hemosiderin deposition may contribute to the epileptogenic potential of cavernomas.9
Cerebral Cysticercosis
Neurocysticercosis currently represents a major public health problem in developing countries of Latin America, Asia, and Africa, as well as in industrialized nations with a high immigration rate from endemic areas.37,133 As the number of immigrants entering the United States from Latin America has increased, so has the incidence of neurocysticercosis. More than 1,000 new cases are being diagnosed in the United States annually, and large neurocysticercosis case series have been reported from California, Texas, Oregon, Chicago, and New York.38,119,128,137,144 Seizures are the most common neurologic symptoms associated with neurocysticercosis, occurring in up to 70% of symptomatic patients. Neurocysticercosis might lead to chronic epilepsy either due to calcified lesions or by triggering epileptogenesis at distant sites (Fig. 4). In a recent study, out
of 512 patients evaluated for intractable epilepsy in an endemic area in Brazil, isolated cysticercosis was found as the cause only in eight patients (1.6%). However, the authors observed calcified cysticercus lesions more frequently in patients with hippocampal sclerosis, raising suspicions of dual pathology.146
of 512 patients evaluated for intractable epilepsy in an endemic area in Brazil, isolated cysticercosis was found as the cause only in eight patients (1.6%). However, the authors observed calcified cysticercus lesions more frequently in patients with hippocampal sclerosis, raising suspicions of dual pathology.146
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


