CHAPTER 215 Lipomyelomeningocele
Johnson first described a lipomyelomeningocele (LMM) in 1857,1 although it was not until 1971 that Rogers and colleagues first introduced the term lipomyelomeningocele in their series of spinal lipomas.2 Since its original description, several authors have subsequently reported on their experience with this very common form of occult spinal dysraphism.3,4 LMM occurs in approximately 1 in every 4000 births in the United States, with a female-to-male prevalence ratio of 1.5 : 1.5 The classification, embryology, diagnosis, and surgical management of LMM are outlined in this chapter.
Definition and Classification
The literature regarding LMM is very confusing. The term lipomyelomeningocele has mistakenly been grouped into the spectrum of all lipomas of the lumbosacral spine and is often misleading because it implies herniation of neural elements through a spina bifida defect into a meningeal sac. Unlike myelomeningoceles or meningoceles, however, there is neither neural nor meningeal tissue found outside the spinal canal. The neural elements remain within the spinal canal, and there is typically an associated subcutaneous lipomatous mass that protrudes through a midline bony defect, displaces the dura, and infiltrates and tethers the spinal cord.5,6 In 1982, Chapman classified three anatomic variants of LMM according to the relationship of the lipoma–spinal cord interface: dorsal, caudal, and transitional.7
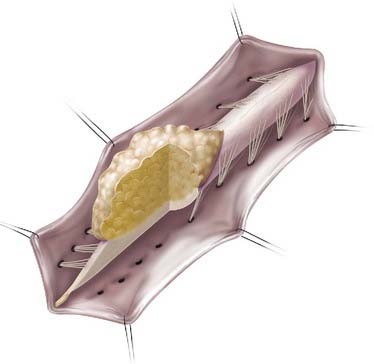
Lipomas that insert onto the dorsal surface of the conus medullaris are classified as dorsal-type LMMs (Fig. 215-1). In this variant there is typically a substantial subcutaneous fat component attached to the underlying spinal cord via a fibrolipomatous stalk of varying thickness. The posterior half of the lower spinal cord at the site of fusion of the neural fold is unfused (i.e., partial dorsal myeloschisis is present), and the stalk attaches directly to the exposed alar and basal cord regions, just posterior and medial to the posterior columns and dorsal to the central canal.8 The lipoma can be located either in the midline (68%) or eccentrically (32%) and can extend into the central canal and expand it for variable distances.9 Along the lateral interface of the attachment of the lipoma to the spinal cord, the dura and pia are fused. Sensory nerve roots emerge just anterior to this lateral line of fusion, which can be traced circumferentially. There is often no evidence of leptomeninges posteriorly, and no sensory or motor roots are found within the actual substance of the lipoma.
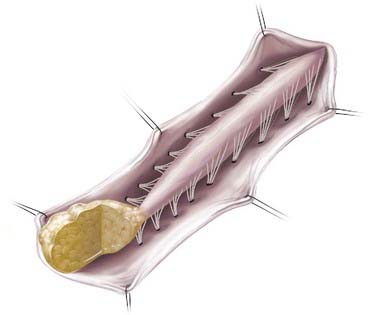
In caudal- or terminal-type LMM, the lipoma–spinal cord interface is located at the caudal end of the conus, almost like a continuation of the spinal cord itself (Fig. 215-2). The remaining lipomatous mass may then lie entirely within the spinal canal or extend dorsally through a defect in the dura and bone into the subcutaneous space. The fatty tumor may either replace or surround the filum terminale, or a separate filum may lie anteriorly. The nerve roots may lie ventral to the lipoma or pass through the mass itself. A caudal-type LMM is often asymmetrical and involves more of the cord and nerve roots on one side, and unlike the discrete lipoma-cord interface of a dorsal-type LMM, the lipoma-cord interface in a caudal-type LMM is diffuse.8
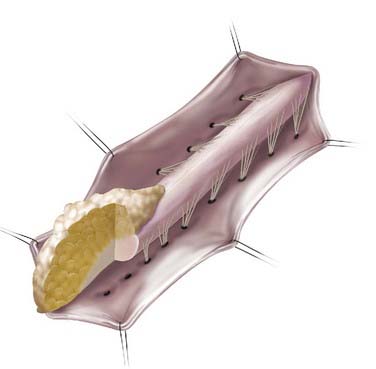
In addition to the dorsal and caudal LMM forms originally described by Chapman, transitional forms between the two variants may exist (Fig. 215-3). Frequently in these cases, the more cephalad portion is of the dorsal type with posterior rootlets emerging from the cord just ventral to the line of fusion of the lipoma to the cord and leptomeninges. Caudally, however, as the line of fusion is displaced ventrally, nerve roots emerge from the anterolateral portion of the lipoma mass. Magnetic resonance imaging (MRI) can often be helpful in defining the type and may be useful in determining the operative approach. The transitional form is considered by some to simply represent a continuum between the other two forms.7,8,10,11
Embryology
Congenital lumbosacral lipomas develop as a result of disturbances in the precise sequence of events encompassing the embryologic stages of primary and secondary neurulation, which occur between postovulatory day (POD) 25 and 48.8 Closure of the posterior neuropore marks the completion of the first phase (primary neurulation) of neural tube formation, at which point the fetal spine is covered by ectoderm but the lumbar, sacral, and coccygeal segments have not yet developed.12 The caudal end of the neural tube and the notochord combine into a large aggregate of undifferentiated cells in a process called condensation. This caudal cell mass extends to the level of the tail fold and represents a solid neural cord containing neurons, neural crest, glial cells, and ependymal cells. In a process called canalization, a series of small vacuoles within the mass begin to coalesce and enlarge to create an ependymal-lined tube in continuity with the rostral central canal of the previously formed neural tube.13 This process takes place between POD 27 and 29 and leads to formation of the ventriculus terminalis.14,15
The third phase in formation of the caudal neural tube involves regression of the structures previously derived during canalization. During retrogressive differentiation, the embryologic tail disappears, whereas the filum terminale, coccygeal ligament, and ventriculus terminalis of the conus remain. This process is thought to complete by POD 48 to 52.12,13,15–17 Because cell rests within the caudal cell mass have totipotent characteristics, a small clone of lipomatous cells developing in the region would not necessarily inhibit the growth of surrounding structures. The bony vertebrae develop subsequently, and the sacrococcygeal segments also undergo regressive changes to decrease the number of segments originally present.15 As a result, vertebral maldevelopments are found in conjunction with the neural defects arising during this period.
McClone and Naidich proposed that the process of dysjunction, in which the closed neural tube detaches from the cutaneous ectoderm, occurs prematurely in patients with LMM. Consequently, a dorsal cleft is maintained and paraxial mesenchyme tissue has access to the prospective lumen of the neural tube, thereby further preventing its closure. The luminal surface of the ectoderm induces mesenchymal cells to differentiate along a path that results in adipocyte formation, whereas the outer surface of the incompletely closed neural tube causes the mesenchyme to form the piaarachnoid and dura.18,19 Other tissues have been found in the substance of an LMM, including striated muscle, cartilage, nerve cells, ependyma, and even cerebellum.6 Similar to other spinal dysraphisms, a lumbosacral lipoma that tethers the spinal cord at the upper lumbar level probably occurs at a slightly earlier embryologic period than does a lesion that tethers the cord lower down in the spine.8 Several authors have found that patients with LMM can have other associated dysraphic conditions of the spinal cord, including split cord malformation, terminal syringomyelia, neurenteric and intraspinal dermoid/epidermoid cysts, a tight filum terminale, and tethering tracts.20,21
Clinical Findings
Cutaneous Signs
Although patients with LMM can have a spectrum of cutaneous anomalies, infants are seen most often with a cutaneous sign that suggests a potential underlying abnormality. The most common finding (>90% of patients) is a subcutaneous fatty mass covered with skin that is located in or near the midline of the lumbosacral spine (Fig. 215-4). This mass is not tender and appears to be in continuity with the normal subcutaneous tissues. Although the overlying skin is typically intact, LMMs can also be associated with a hairy patch (hypertrichosis), hemangioma, dermal sinus, skin dimple, skin tag, atretic meningocele, or caudal appendage (Table 215-1).20,22 The most superficial portion of the mass typically consists of lobulated and unencapsulated subcutaneous fat, which becomes more fibrous as the deeper structures are approached. The stalk of the fibrous fatty tissue can then be found to penetrate the dura and is associated with the spinal cord, conus, cauda equina, or filum terminale, as described earlier. Additional malformations may also be present, including cloacal exstrophy, bifid uterus, duplicated vagina, anterior sacral meningocele, anomalies of the lower limbs, and imperforate anus.15 It is rare for patients with LMM to have intellectual impairment or associated hydrocephalus, Chiari malformations, or other brain anomalies.15,23,24
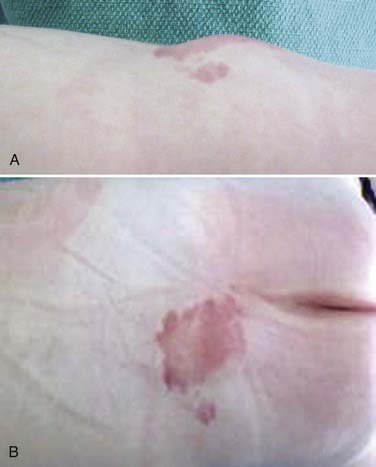
TABLE 215-1 Lumbosacral Cutaneous Manifestations Found in Patients with Lipomyelomeningocele
| SERIES | HOFFMAN ET AL.,20 1985 (N = 97) | KANEV ET AL.,22 1990 (N = 80) |
|---|---|---|
| Soft tissue mass | 97 | 80 |
| Skin dimple | 26 | 14 |
| Hemangioma | 24 | 9 |
| Hypertrichosis | 1 | 11 |
| Skin tag/tail-like appendage | 5 | 6 |
| Atretic or denuded skin patch | 1 | |
| Dermal sinus hypopigmentation | 3 | 3 |
Symptoms
LMMs typically cause tethering of the spinal cord via the associated fibrous subcutaneous lipoma. As infants age, symptoms reminiscent of tethered cord syndrome typically develop, along with any combination of neurologic, orthopedic, and urologic issues. Slightly more than one third of patients will demonstrate an asymmetrical subcutaneous mass with regard to the midline; these patients have a higher incidence of neurological deficits in the lower limb ipsilateral to the side of the mass.25 Symptoms depend on the level of the cord-lipoma interface, as well as patient age at the time of diagnosis, and may be related to several possible causes working independently or in combination.15 Deficits present at birth may become manifested as a result of abnormal development of the spinal cord and nerve roots. The presence of a mass within the confines of the bony spinal canal may also contribute to patient symptoms in more ways than one. Namely, progressive mechanical distortion or ischemia of the neural elements may result from tethering of the neural elements or, alternatively, from a local mass effect with compression of the cord, conus, or cauda equina. Similar to other fatty growths, these tumors can enlarge with age in proportion to normal body growth.26
Although an unknown number of patients may initially be seen with asymptomatic LMMs, it is crucial for clinicians to understand that progressive neurological impairment can develop in patients who at first seem normal. In infants and toddlers, accurate assessment of clinical involvement is very difficult. Yamada demonstrated that spinal cord tethering interferes with normal energy metabolism of the spinal cord, which can lead to ischemia and progressive neural damage.27 In addition, although most patients demonstrate slow loss of function over a period of years, sudden and acute loss of function has also been described. For example, this has been reported in previously asymptomatic women placed in the lithotomy position during pregnancy or in patients with hyperflexion injury from a motor vehicle accident.
Muscle weakness and gait disturbance are often present by the time that the patient starts ambulating. Clinical examination may reveal muscular atrophy, leg length discrepancy, varus or valgus foot deformity of one or both legs, hammertoes or claw toes, scoliosis, mixed deep tendon reflexes (absent, normal, increased), mixed/patchy sensory abnormalities, back pain, or any combination thereof. Abnormal voiding patterns with urinary or fecal incontinence or the presence of urinary tract infections may also be an initial symptom. Although patients can exhibit any of the aforementioned signs and symptoms, infants typically have cutaneous anomalies; older children have orthopedic, neurologic, or urologic symptoms; and adults often have pain.15
Evaluation
Prenatal Diagnosis
LMMs are increasingly being diagnosed in utero as a result of the frequent use of screening antenatal ultrasonography (US).28 Accurate prenatal diagnosis allows appropriate planning for delivery and neonatal care.29 However, because these defects are closed with no communication between fetal spinal fluid and maternal amniotic fluid, amniotic fluid analysis for α-fetoprotein levels is not a reliable diagnostic measure.30–32
Ultrasonography
US has the ability to differentiate among fat, spinal fluid, and spinal cord as a result of their different echogenic properties. Because infants younger than 6 months are skeletally immature with poorly calcified bone, US can image through the spine in these individuals, which can be a very useful tool for defining the attachment of lipomas to the spinal cord.33,34 Several benefits of US include its relative inexpensiveness, ease of use, and lack of need for patient sedation. It is also beneficial in determining spinal cord motion for possible evidence of spinal cord tethering. US is unlikely ever to replace other modalities such as MRI, however, because it is limited in its ability to provide the appropriate preoperative information. In addition, US can be operator dependent and possibly miss subtle lesions unless the user is well experienced.
Plain Radiographs
Plain radiographs can play a beneficial role in the initial assessment of patients suspected of having closed spinal dysraphism; however, as an initial evaluation in most pediatric neurosurgery centers, they are largely being replaced by MRI.35 Anteroposterior and lateral radiographs at the appropriate spinal levels almost always reveal abnormalities such as a dorsal fusion defect in the lamina (bony spina bifida) or widening of the spinal canal.8,36 Varying degrees of agenesis or deformity of the sacrum have also been reported in the literature. Some surgeons prefer to have plain radiographs available for surgical planning. Absence or incomplete calcification of bony elements limits the utility of plain radiographs in children younger than 18 months.37
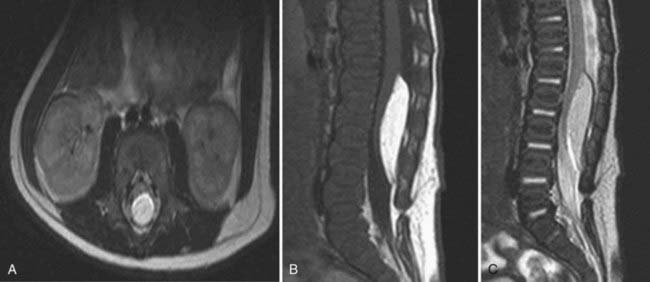
Magnetic Resonance Imaging
MRI is the study of choice for diagnosing and monitoring patients with LMMs.35,38 MRI is especially beneficial in children with congenital disorders because of its capability of three-dimensional imaging and ability to visualize and differentiate neural tissue. T1-weighted imaging provides clear anatomic detail of the spinal cord and filum terminale, which allows visualization of the vertebral level of the conus; the absence, presence, and location of fat within the cord, spinal canal, or filum; and the size of the filum (Fig. 215-5). T2-weighted images allow identification of spinal cord tumors, such as dermoids and epidermoids, as well as the presence of fluid-containing structures such as syringomyelia.36 Performance of MRI to determine spinal cord motion with the patient supine and prone may help one decide whether ventral movement of the cord is adequate and rule out dorsal tethering. The use of gadolinium is typically not required.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


