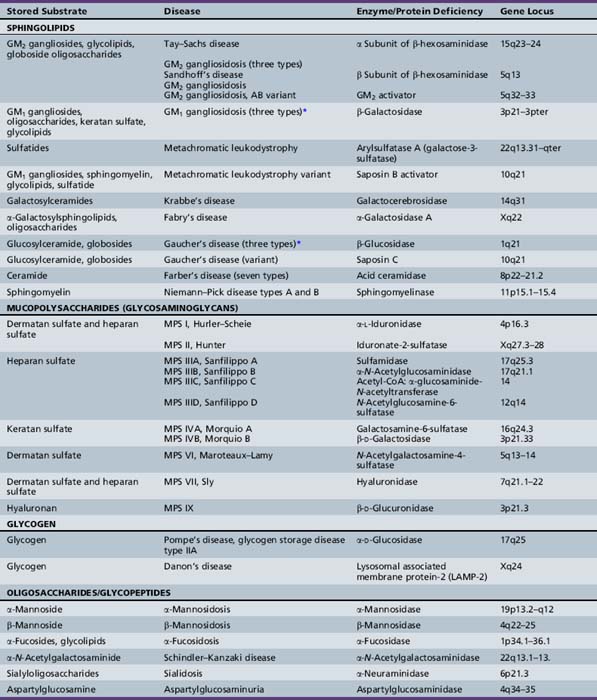
Chapter 36 Lysosomal Storage Diseases
Overview and General Concepts
The lysosomal storage diseases are a clinically heterogeneous group of inborn errors of metabolism that have traditionally been classified according to the biochemical nature of the incompletely degraded macromolecules that accumulate in various tissues. The lysosomal storage diseases are represented by about 50 different clinical entities (Table 36-1). Most of the individual conditions also have an eponymous designation in recognition of the investigator who provided seminal descriptions of the typical manifestations and clinical course of specific variants, often before elucidation of their biochemical or molecular bases.
As a central compartment in the endosomal-lysosomal pathway, the lysosome maintains an acidified milieu enriched with catabolic enzymes to facilitate the degradation of various byproducts of cellular turnover, which are mainly delivered to the lysosome through endocytosis. Phagocytosis and autophagy provide alternative points of substrate entry into the lysosome [Falguières et al., 2009]. These complex substrates may include sphingolipids, glycoproteins, and glycosaminoglycans (mucopolysaccharides). The particular metabolite that builds up (and the ensuing pattern and severity of the associated diseases) is primarily dependent on the substrate tissue-source and the relevant metabolic pathway and transport systems that are compromised. For instance, keratan sulfate is a major constituent of cartilage and the cornea; abnormalities in the turnover of keratan sulfate partly explain the characteristic features of skeletal dysplasia and the corneal opacities associated with the mucopolysaccharidoses [Wegrzyn et al., 2004]. Analysis (through a combination of immune quantification assays and tandem mass spectrometry) of the glycosphingolipid and oligosaccharide profiles in the blood and urine of affected patients may prove useful; as surrogate markers of disease activity, they may enable early diagnosis and the monitoring of clinical responses with directed therapies (such as enzyme replacement or substrate synthesis inhibition) [Parkinson-Lawrence et al., 2006].
The enzymes and transport or integral membrane proteins that are responsible for facilitating the intralysosomal metabolism of these substrates are formed within the endoplasmic reticulum and subsequently refined (through distinct post-translational mechanisms) by the Golgi apparatus [Eskelinen et al., 2003]. The uptake of newly synthesized enzymes by the lysosome, as opposed to other cellular organelles such as the peroxisome or mitochondria, is achieved largely through the mannose 6-phosphate (M6P) receptor pathway. Thus, phosphorylation of the mannose residues of the complex carbohydrate side-chains of the various relevant proteins is an essential step in its maturational processing [Urayama et al., 2004]. Elucidation of this pathway of lysosomal protein delivery, including uptake by the mannose receptors, was critical in the development of enzyme therapy for the lysosomal storage diseases [Grabowski and Hopkin, 2003].
Absent or defective hydrolytic activity within the lysosome, which can come about as a consequence of mutations within the specific encoding gene (for the functional enzyme or its activator or co-factor), is the most common cause of disease (Table 36-2) [Ballabio and Gieselmann, 2009]. For example, deficient glucocerebrosidase activity, as in Gaucher’s disease, results in the accumulation of its substrate, the glycosphingolipid glucosylceramide. Sphingolipid co-factors called saposins facilitate the interaction between the water-soluble glycoprotein enzymes and their lipid-soluble substrates [Matsuda et al., 2007]. Mutation of the saposin C gene is associated with the incomplete metabolism of glucosylceramide in vivo and a rare condition that has clinical features that overlap with the chronic neuropathic form of Gaucher’s disease (type 3 disease). Unlike other lysosomal hydrolases, which rely on the M6P receptor pathway, glucocerebrosidase is targeted to the lysosome via LIMP-2 [Reczek et al., 2007]; mutations of the encoding gene for LIMP-2 have been associated with a progressive myoclonic epilepsy and nephrotic syndrome [Balreira et al., 2008].
Table 36-2 Illustrative List of Underlying Causes
| Etiology | Disease (Examples) |
|---|---|
| Single hydrolytic enzyme deficiency | Gaucher’s disease Tay–Sachs disease Niemann–Pick disease |
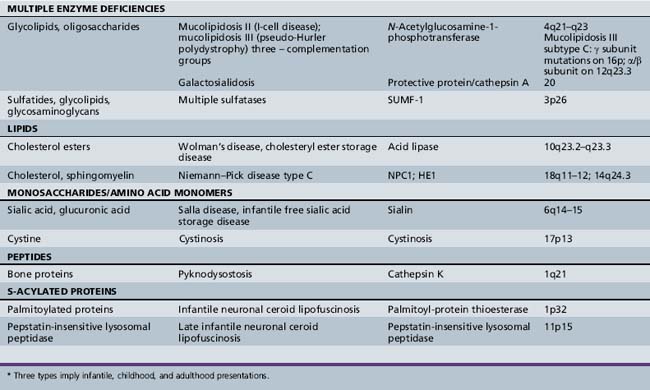
| Single protease enzyme deficiency | Late infantile neuronal ceroid lipofuscinosis (pepinase) |
| Co-factor/activator protein deficiency | Gaucher’s disease variant (saposin) Tay–Sachs disease (GM2 activator) |
| Multiple enzyme deficiencies | Galactosialidosis (protective protein/cathepsin A) Multiple sulfatase deficiency (SUMF-1) I-cell disease (mucolipidosis II) |
| Membrane protein defect | Juvenile neuronal ceroid lipofuscinosis (battenin) |
| Small-molecule transport protein | Cystinosis Sialic acid storage disorders |
| Endocytosis, membrane–vesicle trafficking defect | Mucolipidosis IV Danon’s disease (LAMP-2) Niemann–Pick disease type C |
Other lysosomal disorders result from a defective protein involved in substrate transport (e.g., Niemann–Pick disease type C) or vesicle fusion (e.g., Danon’s disease) [Dierks et al., 2009; Ruivo et al., 2009]. Additional defects of lysosomal transport (e.g., cystinosis, sialic acid storage disorders) involve a dysfunction of intracellular membrane transporters that mediate the movements of the hydrolyzed products outside of the lysosome for their final excretion [Ruivo et al., 2009]. With respect to pathogenesis, cellular toxicity is hypothesized to occur when the lysosomal substrate burden has reached a critical threshold and presumptively triggers a cascade of downstream events that ultimately lead to cell death and the development of disease-specific complications. These consequent mechanisms of disease for most of the individual lysosomal storage diseases remain to be fully clarified.
There is increasing recognition that, besides mechanical forces, other pathologic factors (e.g., inflammation and apoptosis) may play contributory roles in the evolution of the disease phenotype. For instance, in the GM2-gangliosidoses, several markers of an inflammatory response have been found to be elevated and may partly explain the neurodegenerative features encountered in this group of diseases [Jeyakumar et al., 2002, 2003]. Studies have also suggested that certain lysosomal proteases may play active roles in the apoptotic execution process and can act as mediators of programmed cell death [Tardy et al., 2004]. Defects of autophagy are also being recognized as having a role in the pathogenesis of several lysosomal storage diseases [Settembre et al., 2008]. Oxidative damage and cytotoxic cell involvement has been suggested as a factor in the neuronal pathogenesis of mucopolysaccharidosis (MPS) type III [Villani et al., 2009]. Recent studies involving the animal model of mucopolysaccharidosis type I suggest that the absence of recycled precursors results in major shifts in the energy utilization of the cells [Woloszynek et al., 2009]. This finding susggests that lysosomal storage disease may be characterized as diseases of deficiency as well as overabundance (lysosomal storage).
The majority of genes that encode lysosomal proteins are ubiquitously expressed, and each of the various lysosomal storage disease subtypes is characterized by multisystemic involvement. There is wide heterogeneity in the clinical presentation of each entity among individually affected patients, and the concordance between genotype and phenotype is often imperfect even among siblings [Wilcox, 2004]. Disease manifestations may be evident prenatally or at any time from birth to adulthood. Certain lysosomal storage diseases present with nonimmune hydrops fetalis, and the failure to establish causality before the child’s death can lead to potential recurrence in future pregnancies among couples at risk [Staretz-Chacham et al., 2009]. On the other hand, Gaucher’s and Fabry’s diseases may not be associated with prominent symptoms during childhood, and the diagnosis can be missed because the patients have either a mild or an atypical course. This is true especially when the family history is uninformative. To some extent, the variable disease severity encountered between the individual patients has been attributed to different mutations that produce a protein with either no functional activity (in severe cases) or some residual enzyme activity (as observed in those with later onset of clinical expression). Because most lysosomal storage diseases are autosomal-recessive disorders, heterozygotes (or carrier individuals with a single defective gene copy) often have sufficient enzyme activity generated by their other (normal) allele. Carriers for these conditions do not exhibit evidence of tissue storage or suffer from the relevant disease that segregates in the family.
There are at least three disorders, Fabry’s and Danon’s diseases and Hunter’s syndrome (MPS II), that are transmitted as X-linked traits. Although females who are carriers of the trait for Hunter’s syndrome do not appear to develop clinical problems related to the presence of a mutant protein, a proportion of females who are carriers of Fabry’s disease or Danon’s disease may experience disease-related complications, which in a few cases can be as severe as those found in classically affected males. The variable expression in these carrier females has been partly attributed to lyonization (i.e., the random inactivation during early embryogenesis of one of the two X chromosomes). Lyonization results in varying proportions of the mutant gene product in different organ systems [Orstavik, 2009]. Skewed lyonization may be seen in females with an X:autosome translocation. In these cases, there is inactivation of the intact X chromosome. If the X chromosome that is attached to an autosome remains functional and if it happens to bear a gene mutation, disease expression may be unmasked. In addition, rare cases have been reported of females with Hunter’s syndrome who also happen to have Turner’s syndrome (45,X), wherein the single X chromosome that is present bears a mutation of the gene leading to the enzyme (iduronate-2-sulfatase) deficiency.
Although the individual disorders are infrequent to rare and considered orphan disorders by drug regulatory agencies, the lysosomal storage diseases have a combined prevalence of about 1 in 5000 to 8000 [Pinto et al., 2004]. This relatively high occurrence has prompted investigations of the feasibility of targeted screening [Meikle et al., 2004; Zhang et al., 2008]. The rationales given for these programs include:
Table 36-3 Diagnosis and Therapeutic Options for the Lysosomal Storage Disorders
| Disease* | Diagnostic Method† | Treatment‡ |
|---|---|---|
| Tay–Sachs disease (B variant) | Enz; Mol; Prenat (Enz/Mol) | SRT (in clinical trials) |
| GM2 gangliosidosis (three types) | ||
| Sandhoff’s disease (O variant) | Urine oligo; Enz; Mol; Prenat (Enz) | Palliative |
| GM2 gangliosidosis | ||
| GM2 gangliosidosis, AB variant | Func; Mol; Prenat (Mol) | Palliative |
| GM1 gangliosidosis (three types) | Enz; Mol; Prenat (Enz) | Palliative |
| Metachromatic leukodystrophy | Enz; Mol; Prenat (Enz) | HSCT/BMT |
| Metachromatic leukodystrophy variant (saposin B deficiency) | Func/Subs; Mol; Prenat (Mol) | Palliative |
| Krabbe’s disease | Enz; Mol; Prenat (Enz) | HSCT/BMT |
| Fabry’s disease | Enz/Subs; Mol; Prenat (Enz/Mol) | ERT |
| Gaucher’s disease types 1, 2, and 3 | Enz; Mol; Prenat (Enz) | ERT, primarily for types 1 and 3; SRT |
| Gaucher’s disease (saposin C variant) | Func/Subs; Mol; Prenat (Mol) | Palliative |
| Farber’s disease (seven types) | Enz; Mol; Prenat (Enz) | Palliative |
| Niemann–Pick disease types A and B | Enz; Mol; Prenat (Enz) | ERT (in clinical trials primarily for type B) |
| MPS I, Hurler–Scheie | Urine GAG; Enz; Mol; Prenat (Enz) | ERT; HSCT/BMT |
| MPS II, Hunter | Urine GAG; Enz; Mol; Prenat (Enz) | ERT (in clinical trials) |
| MPS IIIA-D, Sanfilippo A to D | Urine GAG; Enz; Mol; Prenat (Enz) | Palliative |
| MPS IVA, Morquio A | Urine GAG; Enz; Mol; Prenat (Enz) | Palliative |
| MPS IVB, Morquio B | Urine GAG; Enz; Mol; Prenat (Enz) | Palliative |
| MPS VI, Maroteaux–Lamy | Urine GAG; Enz; Mol; Prenat (Enz) | ERT (in clinical trials) |
| MPS VII, Sly | Urine GAG; Enz; Mol; Prenat (Enz) | Palliative |
| MPS IX | Enz; Mol; Prenat (none reported) | Palliative |
| Pompe’s disease, glycogen storage disease IIA | Urine oligo; Enz; Mol; Prenat (Enz) | ERT (in clinical trials) |
| Danon’s disease | Mol; Prenat (Mol) | Palliative |
| α-Mannosidosis | Urine oligo; Enz; Mol; Prenat (Subs) | HSCT/BMT |
| β-Mannosidosis | Urine oligo; Enz; Mol; Prenat (Enz) | HSCT/BMT |
| α-Fucosidosis | Mol; Prenat (Mol) | HSCT/BMT |
| Schindler–Kanzaki disease | Urine oligo; Enz; Mol; Prenat (Enz) | Palliative |
| Sialidosis (mucolipidosis I) | Enz; Mol; Prenat (Enz) | Palliative |
| Aspartylglucosaminuria | Urine oligo; Enz; Mol; Prenat (Enz) | Palliative |
| Mucolipidosis II (I-cell disease) | Urine oligo; Mul Enz; Mol; Prenat (Enz) | Palliative; BMT (1 case) |
| Mucolipidosis IV | Mol; Prenat (Mol) | Palliative |
| Galactosialidosis (protective protein/cathepsin A deficiency) | Urine oligo; Mul Enz; Mol; Prenat (Mul Enz) | Palliative |
| Multiple sulfatases | Urine GAG; Mul Enz; Mol; Prenat (Enz) | Palliative |
| Wolman’s disease, cholesteryl ester storage disease (acid lipase deficiency) | Enz; Mol; Prenat (Enz) | Palliative |
| Niemann–Pick disease type C | Func/Subs; Mol; Prenat (Func/Subs) | SRT (in clinical trials) |
| Salla disease, infantile free sialic acid storage disease | Subs; Mol; Prenat (Subs) | Palliative |
| Cystinosis | Func/Subs; Mol; Prenat (Func/Subs) | Cysteamine |
| Pyknodysostosis | Enz; Mol; Prenat (none reported) | Palliative |
| Neuronal ceroid lipofuscinosis | Enz; Mol; Prenat (Enz/Mol for certain subtypes) | Palliative |
† Enz, enzyme assay; Func, functional assay (see text); GAG, glycosaminoglycans; Mol, molecular/DNA/gene defect analysis; Mul Enz, multiple enzyme in either cultured cells/amniotic fluid or serum media; Oligo, oligosaccharides; Prenat, prenatal diagnosis available; Subs, substrate analysis (see text).
‡ BMT, bone marrow transplantation; ERT, enzyme replacement therapy; HSCT, hematopoietic stem cell transplantation; SRT, substrate reduction therapy.
Several advances in the understanding of lysosomal storage diseases have resulted from investigations of animal disease models, which either have arisen spontaneously or have been generated by the application of recombinant genetic techniques (primarily involving mice) [Suzuki et al., 2003]. Numerous insights into the role of certain metabolites in embryonic and fetal development and their functions in various organs have been obtained from experiments involving these different animal models, which have also been useful in the development and preclinical testing of new putative therapies [Desmaris et al., 2004; Haskins, 2009]. Although most mouse disease models are genetically authentic and mimic the human phenotype, several reproduce only some aspects or may have no clinical manifestations at all (e.g., the Tay–Sachs disease knockout mouse) [Elsea and Lucas, 2002].
On the therapeutic front, industrial-scale production of recombinant human enzymes (through manipulation of mammalian cells in culture) has enabled the introduction of enzyme replacement therapy for several disorders, including Gaucher’s, Fabry’s, and Pompe’s (glycogen storage disease II) diseases, and Hurler–Scheie (MPS I H/S), Hunter’s (MPS II) and Maroteaux–Lamy (MPS VI) syndrome [Pastores, 2003; Pastores and Barnett, 2005]. In diseases primarily associated with central nervous system (CNS) degeneration (e.g., Tay–Sachs disease and Niemann–Pick disease type C) and for which protein delivery across the blood–brain barrier represents a major obstacle, trials with substrate reduction therapy have been undertaken [Platt and Lachmann, 2009]. The rationale for this approach is based on the following: in cases wherein mutant cells express residual enzyme activity, metabolic homeostasis may be restored or maintained by limiting the amount of intralysosomal substrate accumulation through inhibition of the amount of precursor material that is synthesized [Pastores and Barnett, 2003; Platt and Jeyakumar, 2008]. In Gaucher’s disease, substrate reduction therapy is feasible with the use of the imino sugar miglustat (which inhibits the activity of ceramide-specific glucosyltransferase, the rate-limiting enzyme involved in the biosynthesis of glycosphingolipids).
Before enzyme replacement therapy and substrate reduction therapy, bone marrow transplantation was undertaken in selected cases (see Table 36-3) [Prasad and Kurtzberg, 2008]. The high procedure-related morbidity and mortality risks associated with bone marrow transplantation (e.g., from graft-versus-host disease or infections) preclude its general application, even in cases in which clear therapeutic gains could be established in the successfully engrafted patients. The use of hematopoietic or mesenchymal stem cells and less intensive (i.e., nonmyeloablative) conditioning regimens is being examined as a means to improve clinical outcome and to minimize procedural risks [Prasad and Kurtzberg, 2008].
Another novel approach, termed enzyme enhancement therapy, is also under investigation. Enzyme enhancement therapy is a potential treatment strategy for diseases resulting from a defective enzyme associated with a mutation that leads to its misfolding and rapid degradation [Desnick, 2004]. In these instances, the use of an agent that acts as a chaperone to stabilize the mutant enzyme may help restore its function. Proof of principle for this strategy has been demonstrated in Fabry’s disease, for which the regular intravenous administration of galactose has led to the resolution of cardiomyopathy and improvement in the associated hemodynamic changes in one patient [Frustaci et al., 2001]. Currently, clinical trials with a pharmacologic chaperone are being undertaken in Gaucher’s and Fabry’s diseases [Pastores and Sathe, 2006].
Gene therapy is also being explored as an option, although most studies to date have only been with animal (primarily mouse) models, except for Gaucher’s disease, for which clinical trials have been performed [Caillaud and Poenaru, 2000; Ellinwood et al., 2004; Eto et al., 2004; Gieselmann et al., 2003b; Shen et al., 2004; Sly and Vogler, 2002]. Unfortunately, gene therapy performed in the patients with Gaucher’s disease resulted in only transient expression of the functional enzyme [Cabrera-Salazar et al., 2002]. Although experiments in animal models provide a foundation for translational studies in humans, the increased size and complexity of the human brain present particular challenges that may not be clarified by studies in the mouse models. Thus, studies in large animal models are increasingly preferred [Haskins, 2009]. Additionally, immunological rejection of the vector and viral products may lead to adverse outcome; ways to address these problems will be needed, to reduce procedure-related risks and promote long-term gene expression. Furthermore, the CNS and synovial joints are sites that can be challenging to treat with systemic gene therapy, and may require direct approaches for delivery of the relevant gene. For most lysosomal storage diseases, the mainstay of treatment has been palliative or supportive care. Treatment of the secondary disabilities (e.g., seizures, sensory impairment, and behavioral, sleep–wake cycle, or communication problems) can have a positive impact on the patient’s quality of life and may help address some sources of parental frustration. Genetic counseling of affected individuals and their relatives is also an important component of comprehensive care of the patient and family.
Sphingolipidoses
The sphingolipidoses are several disorders characterized by abnormalities in the metabolism of multiple glycolipid substrates that form a component of myelin or of lipid rafts within membranes (Figure 36-1). Most of the clinical variants in this group are associated with neurodegenerative features.
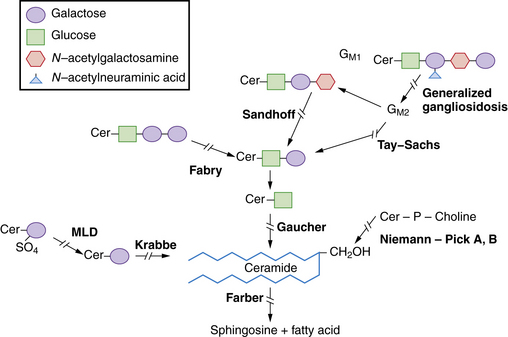
The myelin sheath is an extended, modified plasma membrane that, in the CNS, represents an extension of oligodendroglial cell processes. Examination of normal myelin with polarized microscopy and radiographic diffraction has exhibited a protein-lipid-protein-lipid-protein structure. Chemical investigations have determined that the lipid layers are composed of a bimolecular layer of hydrocarbon chains, cholesterol, phospholipids, and glycolipids (primarily galactocerebroside and sulfatide). The glycolipids are amphiphilic and have a hydrophobic ceramide moiety that acts as a membrane anchor and a hydrophilic, extracellularly oriented oligosaccharide chain. Rather than diffuse distribution in the cell membrane, current studies support the localization of glycosphingolipids, cholesterol, and certain proteins in specialized patches of membranes referred to as lipid rafts. Lipid rafts play key roles in intracellular transport, protein-sorting, and signal-transduction processes. Disturbed distribution of raft lipids and consequently protein subcellular localization due to a gridlock in the endocytic pathway, as seen in certain lysosomal storage diseases, is believed to lead to cell and organ functional impairments [Saravanan et al., 2004; Walkley and Vanier, 2009]. In addition, abnormalities of myelin membrane lipid composition result in the loss of membrane stability and its degeneration.
The degradation of glycosphingolipids takes place in the lysosomes through a stepwise action of specific acid hydrolases. Several of these enzymes (such as β-glucosidase, β-galactocerebrosidase, arylsulfatase A, and acid ceramidase) also require the assistance of nonenzymatic glycoprotein co-factors, the so-called sphingolipid activator proteins (or saposins). Examination of tissue sections obtained from affected individuals with glycosphingolipid storage disease typically reveals membranous cytoplasmic bodies, which represent lipid material enclosed by the lysosomal membranes [Abramovich et al., 2001].
Studies in animal models of Sandhoff’s disease and Niemann–Pick disease type A revealed elevated levels of phosphatidylcholine surfactant in lung tissue [Buccoliero et al., 2004]. Subsequent studies have confirmed these findings in human patients with Sandhoff disease, Gaucher disease type I, and sialidosis type I [Buccoliero et al., 2007]. These observations suggest that changes in phospholipid levels and composition in lung surfactant might be a general feature of sphingolipid storage diseases and may in part be responsible for the increased susceptibility of these patients to respiratory infections and lung disease.
Gangliosidoses
Ganglioside (primarily GM2 and GM3) accumulation has also been found to occur secondarily in other disorders, such as the mucopolysaccharidoses [Walkley and Vanier, 2009]. In the mucopolysaccharidoses, the primary storage of glycosaminoglycans is believed to lead to inhibition of the activity of several ganglioside-degrading lysosomal enzymes. In other disorders (e.g., Niemann–Pick disease type C), secondary ganglioside accumulation is believed to result from a disruption in retroendocytic movement of the substrate and does not appear to reflect simply the nonspecific changes in tissue cellularity (arising from neuronal loss and gliosis) [Walkley and Vanier, 2009].
Neuronal cells that have GM2 ganglioside storage exhibit meganeurite formation (i.e., axonal hillock enlargement) and ectopic dendritogenesis (i.e., the sprouting of new synapse-covered dendritic neurites at the axon hillock) [Walkley and Vanier, 2009]. Axonal spheroid formation (or neuroaxonal dystrophy) is also often noted. The spheroids occur as focal enlargements of various sizes scattered along myelinated and unmyelinated axons in the gray and white matter. In contrast to neuronal cell bodies, which contain characteristic storage material, spheroids consist of collections of multivesicular and dense bodies, mitochondria, and other organelles that would normally be found being transported along axons [Walkley and Vanier, 2009]. These observations suggest that the development of spheroids may involve defective endocytic trafficking within axons.
GM1 Gangliosidoses
Deficient β-galactosidase activity leads to the incomplete metabolism of several other substrates, including galactose-containing glycoproteins, N-acetylgalactosamine, lactose, and keratan sulfate (a glycosaminoglycan). Phenotypic differences likely result from varying activities of the mutant β-galactosidase enzyme against its various substrates, which may explain the dysmorphic facial features reminiscent of the mucopolysaccharidoses in the early infantile form of GM1 gangliosidosis and the predominance of skeletal disease in Morquio’s syndrome type B (due to allelic mutations) [Brunetti-Pierri and Scaglia, 2008]. Allelic mutations involve distinct sequence alterations of the same gene; the phenotype encountered may be different because the specific underlying gene defects involve distinct domains of the protein that subserve different functions. Studies in the mouse model of GM1 gangliosidosis have found that GM1 storage leads to several cellular changes (including activation of the unfolded protein response pathway), the upregulation of BiP and CHOP (C/EBP homologous transcription factor), and the activation of JNK2 and caspase 12, which lead to neuronal apoptosis [Tessitore et al., 2004].
Histologic examination of the brain of two infants with GM1 gangliosidosis revealed a marked decrease in the number of oligodendrocytes and myelin sheaths [van der Voorn et al., 2004]. An immunohistochemical decrease in proteolipid protein and a more profound deficiency of myelin basic protein were also found; these observations indicate that the brain lesions may not simply result from a delay or arrest in myelination, but are due to a “dying-back” oligopathy. In addition, amyloid precursor protein–immunoreactive aggregates were observed in proximal axons and meganeurites, as well as in white matter axons. These data suggest that the myelin deficit results from a loss of oligodendrocytes and abnormal axoplasmic transport, consequent to the massive neuronal storage of GM1 gangliosides.
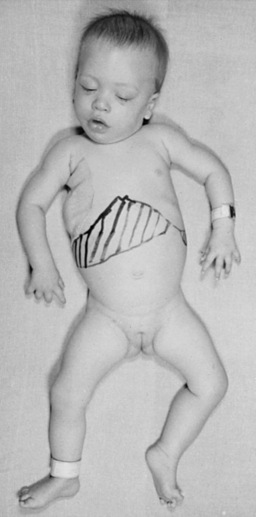
The late infantile form (with onset usually between 12 and 18 months of life) often manifests with gait disturbances and frequent falls. There is usually no distinctive facial dysmorphism, and the liver and spleen are not enlarged. Skeletal deformities, such as hypoplasia of the acetabula and proximal deformity of the metacarpal bones, may be found on radiographic examination but are usually milder than those noted in the early infantile form of GM1 gangliosidosis. Patients develop seizures and spastic quadriparesis, with prominent pseudobulbar signs (i.e., drooling and dysphagia) (Figure 36-2). Death is usual between the ages of 3 and 10 years. The absence of peripheral nerve involvement (i.e., normal conduction velocities) and normal cerebrospinal fluid findings help distinguish this condition from metachromatic leukodystrophy and Krabbe’s disease.
In the juvenile or late-onset form, disease symptoms usually develop in late childhood or adolescence but may also occur as late as the third or fourth decade of life. Delayed onset is partly related to the presence of residual enzyme activity, which in one patient was reported to be associated with homozygosity for R521C mutation in the β-galactosidase gene [Silva et al., 1999]. This clinical variant has been observed in different ethnic groups but appears to be prevalent among the Japanese. Affected individuals have a protracted clinical course characterized by dysarthria and extrapyramidal signs (especially dystonia). Pathologic studies have revealed marked GM1 ganglioside storage in the basal ganglia. Intellectual impairment is slight to moderate.
Mutation analyses have revealed broad heterogeneity, with 102 β-galactosidase gene defects described to date [Brunetti-Pierri and Scaglia, 2008]. Certain gene defects tend to be common in particular subtypes; for instance, the R482H and R208C are often seen in infantile cases, R201C in the late infantile/juvenile patients, and I51T among rare adult/chronic cases. In contrast to the first two mutations, the last two defects are associated with moderate catalytic activity, consistent with expectations for the resultant phenotype. The R59H has been reported to be prevalent among Brazilians, Iberian, and Roma patients with GM1 gangliosidosis.
Only symptomatic treatment is available. In preclinical studies, substrate reduction therapy using two related imino sugars revealed that NB-DGJ led to an optimal response (in terms of survival and fewer side effects in a mouse model of GM1 gangliosidosis), when compared to treatment with NB-DNJ (miglustat). However, functional improvement was greater with NB-DNJ, probably due to the greater impact of NB-DNJ on CNS inflammation [Elliot-Smith et al., 2008].
GM2 Gangliosidoses
The GM2 gangliosidoses include several variants, all of which are associated with the neuronal storage of the monosialoganglioside GM2, resulting in the formation of meganeurites (i.e., distorted and ballooned neurons) and ectopic dendritogenesis. Primary accumulation of GM2 gangliosides occurs with mutations involving the genes encoding the α (Tay–Sachs disease) or β (Sandhoff’s disease) subunit of hexosaminidase A or the GM2 activator protein (in the AB variant) [Fernandes Filho and Shapiro, 2004]. The hexosaminidase A enzyme (with a molecular mass of approximately 100 kDa) is a trimer consisting of one α and two β subunits, encoded by structural genes on different chromosomes (see Table 36-1). Mutations of the β subunit also lead to deficiency of hexosaminidase B (a tetrameric homopolymer of β subunits).
GM2 ganglioside storage is also seen in the so-called B1 variant, resulting from an altered substrate specificity of hexosaminidase A, wherein the mutated enzyme retains the ability to degrade the artificial substrate (used in most diagnostic assays), but not the sulfated or natural substrate in vivo. Clinical and imaging findings are congruent with those reported for Tay–Sachs and Sandhoff’s diseases [Grosso et al., 2003]. The highest incidence (approximately 3.1 per 100,000 live births) for the B1 variant of GM2 gangliosidoses has been described in Portugal, which has been suggested as the point of origin of a founder mutation (R178H) seen in Brazilian patients [Tutor, 2004].
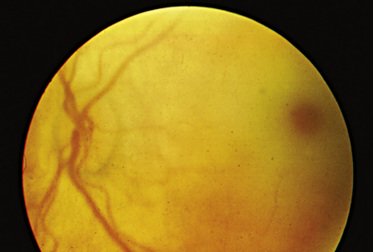
The “classic” infantile form of Tay–Sachs disease is named after a British ophthalmologist (Warren Tay) and an American neurologist (Bernard Sachs); both provided initial descriptions (in the 1880s) of the typical disease manifestations. Affected infants with Tay–Sachs disease have psychomotor deterioration and hyperacusis together with axial hypotonia, bilateral pyramidal signs, and blindness (associated with persistent pupillary responses) (Figure 36-3). The macular cherry-red spot is a characteristic hallmark of the disease (Figure 36-4). Head control is poor, and the size of the head increases markedly in the second year of life from enlargement of the brain (megalencephaly) and not because of tension hydrocephalus. Ultimately, with loss of the neurons and further gliosis, the ventricles may appear enlarged. Affected children become spastic and cachectic. Death usually occurs between 3 and 5 years of age. As patients approach the terminal stages, generalized tonic-clonic and minor motor seizures can occur. The patients do not have any of the following signs: dysmorphic features, visceromegaly, skeletal deformities, and signs of peripheral nerve involvement. These observations are useful in the delineation of Tay–Sachs disease from other lysosomal storage diseases associated with neurodegenerative features (Box 36-1).

Box 36-1 Clinical Manifestations Reported in Patients with Lysosomal Storage Disorders




























































































































* Conductive, sensorineural, or a combination, with involvement of cochlear and CNS dysfunction.
† Describes the normal red macula surrounded by a pale retina, reflecting storage material in the perifoveal ganglion cells.
‡ Refers to constellation of radiologic findings, including macrocephaly, thickened calvaria, J-shaped sella turcica, spatulate ribs, bullet-shaped phalanges that are shortened, rounding and anterior beaking of the vertebral bodies (especially in the lumbar region), and flaring of the iliac wings.
In the general population, the incidence of Tay–Sachs disease has been estimated at 1 in 112,000 live births. Tay–Sachs disease used to be prevalent (1 in 3900 live births) among Ashkenazi Jews, but this situation has been addressed successfully through targeted screening programs. The success of targeted screening has prompted extension of the program to other disorders that are also common in Ashkenazi Jewish individuals, such as Gaucher’s disease, Niemann–Pick disease, and mucolipidosis IV. These particular disorders were chosen because they are found with increased carrier frequency among the Ashkenazi Jews, in whom causality has been attributed to a limited number of “common” mutations [Zhang et al., 2004].
The later-onset forms of GM2 gangliosidosis (referred to as LOTS, which stands for late-onset Tay–Sachs disease) can manifest in childhood, adolescence, or even adulthood. In contrast to the infantile-onset form, patients with juvenile- or adult-onset disease follow a more protracted course and show no ethnic predilection. Affected individuals develop dysarthria and walking problems, primarily resulting from spastic paraparesis (in the childhood-onset form, between the ages of 3 and 6 years) or proximal muscle weakness (in the adult form, with symptom onset in the teens). Differences in the age at onset and disease course have led to distinction of the childhood-onset form (usually referred to as chronic GM2 gangliosidosis) from the adult-onset variant (or late-onset Tay–Sachs disease) [Maegawa et al., 2006]. The phenotype associated with the B1 variant is similar to that encountered in the childhood-onset form.
Ataxia with cerebellar atrophy is also a prominent disease sign in the later-onset forms, and peripheral neuropathy has been described in a subset of patients [Shapiro et al., 2008]. Tonic-clonic or myoclonic seizures are encountered in some children; psychiatric disturbances (i.e., bouts of psychosis and episodes of depression) are present and may be the initial manifestation of disease, particularly among adult-onset patients [Zaroff et al., 2004]. Consideration of late-onset GM2 gangliosidosis should be made in the differential diagnosis of patients with signs of lower motor neuron and spinocerebellar dysfunction. Vision is not impaired and optic fundi are normal, although intellectual deterioration is frequent. The rate of progression and disease severity usually correlate with the age at onset (presumably determined by the severity of the underlying mutation). Childhood-onset disease often leads to death by 15 years of age, preceded by a period characterized by progressive spasticity, rigidity, and dementia, and ending in the vegetative state. Adult-onset patients may live into their 50s or early 60s. Mutation analyses performed in patients with late-onset Tay–Sachs disease have identified a high prevalence of the G269S gene defect, which is associated with residual hexosaminidase A activity, usually in combination with a null allele typical of classic Tay–Sachs disease [Neudorfer et al., 2005].
The AB variant due to a deficiency of the GM2 activator, which is necessary for the hydrolysis of GM2 gangliosides by hexosaminidase A in vivo, has a clinical phenotype that is indistinguishable from the infantile form of Tay–Sachs disease [Li et al., 2003; Sakuraba et al., 1999]. It is a rare condition associated with normal laboratory test results in assays of hexosaminidase A and B enzyme activity with use of the artificial substrates. Diagnostic confirmation requires highly specialized methods, available only through a few laboratories.
Sandhoff’s disease (O variant), due to mutations of the β subunit of hexosaminidase A, has an age at onset and clinical course characterized by neurologic and ophthalmologic findings that are similar to Tay–Sachs disease. Sandhoff’s disease is a pan-ethnic disorder. A distinguishing feature is hepatosplenomegaly and the presence of N-acetylglucosamine–containing oligosaccharides in urine (as only hexosaminidase B, which is also deficient in Sandhoff’s disease but not in Tay–Sachs disease, is responsible for cleaving the N-acetylglucosamine moiety of globosides) [Sakuraba et al., 2002]. Cardiomyopathy has also been noted occasionally. Death usually occurs between 2 and 4 years of age. There are extremely rare cases of patients with juvenile- or late-onset Sandhoff’s disease [Hendriksz et al., 2004].
The enzymatic basis of GM2 gangliosidosis can be ascertained by assays of total hexosaminidase and hexosaminidase A activity, which can be measured in leukocytes and cultured skin fibroblasts. The biochemical tests can be complemented by analysis of the underlying hexosaminidase A gene mutations. Among the Ashkenazi Jewish population, two null hexosaminidase A mutations (a four-base pair insertion [TATC1278] in exon 11 and a splice site gene defect [IVS12]) account for 96 percent of disease alleles among cases of Tay–Sachs disease [Frisch et al., 2004]. In this same population of patients, an allelic glycine to serine substitution at position 269 of the hexosaminidase A gene (G269S) represents the most common mutation among the adult-onset cases of GM2 gangliosidosis. Among non-Jewish patients, one mutation (IVS9) accounts for 14 percent of disease alleles [Cordeiro et al., 2000]. Pseudodeficiency for hexosaminidase A activity associated with decreased enzyme activity in vitro exists and can be clarified by DNA testing or analysis of enzyme activity with use of the natural rather than the artificial substrate. Two mutations (i.e., R247W and R249W) have been associated with pseudodeficient hexosaminidase A activity [Cordeiro et al., 2000].
Investigations have proved that the infantile cases of Tay–Sachs disease are usually due to a combination of severe deleterious mutations, whereas patients with the later-onset forms often have a combination of one deleterious mutation and a second “milder” mutation associated with residual enzyme activity (based on in vitro mutagenesis and protein expression studies). The most frequent mutation responsible for the B1 variant of GM2 gangliosidosis is R178H [Matsuzawa et al., 2003].
Pathologic brain studies reveal widespread neuronal GM2 ganglioside storage throughout the cortex and central nuclear structures, the spinal cord, and the autonomic ganglia. Evidence of neuronal storage has been established in fetuses as early as 12–22 weeks of gestation. GM2 gangliosides are implicated in synaptogenesis, and it has been speculated that an altered pattern of cellular gangliosides may interfere with normal synaptic transmission [Zhao et al., 2004].
Lyso-derivatives of GM2 gangliosides, which are cytotoxic and may be responsible for inducing the neuronal degeneration, are found to be increased in tissues of patients with GM2 gangliosidoses. When they were initially described, the lysolipids were believed to exert their toxicity through inhibition of protein kinase C. More recently, studies in animal models have found that progressive CNS inflammation (as evident from the expression pattern of several inflammatory markers and cytokines) and an alteration in the integrity of the blood–brain barrier occurred coincidentally with the onset of clinical disease signs [Jeyakumar et al., 2003].
Definitive therapy for GM2 gangliosidoses is not available. Substrate reduction therapy is currently under investigation. This approach is based on the inhibition of glucosylceramide synthesis by the imino sugar miglustat (NB-DNJ), and more recently by the galacto-derivative [Andersson et al., 2004]. Preclinical studies have found that animal models of GM2 gangliosidosis (primarily the Sandhoff mouse) given miglustat had a reduction in tissue storage of GM2 ganglioside, with delayed symptom onset and extended survival [Jeyakumar et al., 1999]. Treatment with NB-DGJ demonstrated greater therapeutic efficacy (specifically, extended life expectancy and increased delay in symptom onset) than NB-DNJ, with none of the side effects, such as weight loss and gastrointestinal tract distress, reported with NB-DNJ [Andersson et al., 2004]. Unfortunately, clinical trials using miglustat did not alter ultimate neurologic prognosis in pediatric and adult patients with GM2 gangliosidosis [Maegawa et al., 2009; Shapiro et al., 2009]. Salutary changes noted in the animal models subjected to bone marrow transplantation have generated renewed interest in cellular approaches to treatment of the GM2 gangliosidoses; potentially in combination with substrate reduction therapy [Jeyakumar et al., 2001].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


