Chapter 26 Malformations of Cortical Development
The classification scheme for MCD followed in this chapter (see Box 21-2) is based on enumeration of the developmental step(s) at which the process was first disturbed, the underlying genes and biological pathways disrupted, and – when more objective data are not available – brain imaging features [Barkovich et al., 1996, 2001b, 2005]. This system classifies MCD into three major groups, as noted above: those resulting from abnormalities of cell proliferation; those resulting from abnormalities of neuronal migration; and those resulting from abnormal cortical organization. The large subset of disorders presenting with abnormal brain size – microcephaly and megalencephaly – are reviewed in Chapter 25. The subset of developmental disorders of the cortex in which alterations in brain size are not the dominant feature are reviewed in the present chapter. These include the primary neuronal migration disorders, lissencephaly (including agyria and pachygyria) and subcortical band heterotopia, cobblestone-type cortical malformations, periventricular and subcortical nodular heterotopia, polymicrogyria (including schizencephaly), and other focal cortical dysplasias.
Embryology
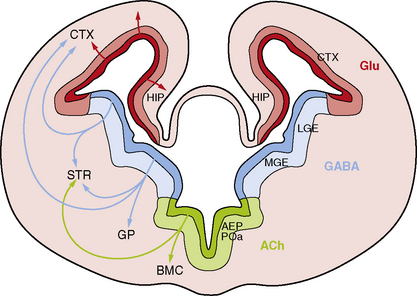
While the major stages of cortical development have been known for decades [Sidman and Rakic, 1982], several recent discoveries have led to a sea change in our understanding of this complex process. The most important of these include recognition that the primary neural progenitor cells and the classic radial glia cells that guide neuronal migration to the cortical plate are the same cells, that the remarkable expansion of the cerebral cortex in humans results in part from neurogenesis in the subventricular zone, and that an important class of cortical neurons originates from ventral brain regions (Figure 26-1).
Classic Studies
By GW5, the rostral forebrain or telencephalon begins as a single layer of proliferative neuroepithelial cells overlying the newly formed lateral ventricles [Kriegstein et al., 2006; Norman et al., 1995; Sidman and Rakic, 1982]. This primitive layer, known as the ventricular zone (VZ), consists of densely packed and morphologically homogeneous cells that are radially oriented and maintain contact with both the ventricular lumen and the pial surface of the brain. Neural progenitor cells or neuroblasts in the VZ have highly mobile nuclei that move up and down within the cytoplasm of the cell, undergoing division when the nuclei are located near the ventricular surface.
During cortical development, the marginal zone contains pioneer neurons known as Cajal–Retzius cells, which express Reelin and other proteins that regulate neuronal migration, cortical lamination, and later cortical organization [D’arcangelo et al., 1995]. The upper part of the marginal zone persists to form layer one of the mature cortex. Subplate neurons participate in development of critical thalamocortical and corticocortical connections, and also express many regulatory proteins before disappearing late in gestation [Del Rio et al., 2000]. The cortical plate grows rapidly, with newly arriving neurons migrating past earlier-generated cells to form progressively more superficial layers. This process of “inside-out” migration ultimately forms a six-layered cortex in which a neuron’s final position is determined primarily by its birth date. The VZ becomes progressively smaller and is eventually replaced by a single layer of ependymal cells that line the lateral ventricles. The SVZ eventually disappears as well, except along the lateral wall of the lateral ventricles, where it persists and continues to provide olfactory and possibly other neurons into adulthood.
Neurogenesis
First, vzRG were found to be primitive multipotential cells that serve as the primary neuronal founder cells, as well as classic radial glia cells [Heins et al., 2002; Malatesta et al., 2000; Miyata et al., 2001; Noctor et al., 2002]. During early stages of cortical development, radial founder cells undergo several sequential symmetric cell divisions to generate additional founder cells. Once some critical number of radial founder cells has been reached – a number that appears to differ between species – the process changes to a form of asymmetric division that generates one vzRG and one immature neuron. The immature neuron then migrates up the radial glia fiber to reach the cortical plate. The daughter cells generated from each radial founder cell are thought to generate a functional neuronal column or radial unit. The radial unit hypothesis [Rakic, 1995, 2000] proposes that the evolutionary expansion in cortical surface area in primates resulted from an increase in the number of founder cells prior to neurogenesis, with each radial unit generating a column of six layers of neurons. This would have the effect of increasing cortical and ventricular surface area without increasing cortical thickness.
Second, neuronal progenitor cells are found in the SVZ, as well as the VZ. The first type discovered were intermediate progenitor cells (IPCs), which divide symmetrically to produce immature neurons [Haubensak et al., 2004; Miyata et al., 2004; Noctor et al., 2004]. However, the outer portion of the SVZ is also populated with nonepithelial radial glia-like (oRG) cells that are able to self-renew and produce neurons, similar to classic vzRG [Hansen et al., 2010]. Both vzRG and oRG cells undergo asymmetric cell division to produce IPCs, which divide one or a few times to produce immature neurons. The two populations of outer SVZ progenitors – oRG and IPCs (both also called transit amplifying cells) – contribute to a massively expanded outer SVZ in humans and produce a majority of neurons destined for the human cortex [Hansen et al., 2010; Haubensak et al., 2004]. Further, most newborn neurons do not migrate directly to the cortex. Rather, they demonstrate several distinct phases of migration, including a phase of retrograde movement toward the ventricle before migration out to the cortical plate [Noctor et al., 2004].
Specification of neuronal cell type occurs very early in the process, which is regulated by expression of a series of proteins. For example, vzRG and probably oRG progenitors express the paired-class transcription factor Pax6, whereas IPCs express the T-domain transcription factor Tbr2 (also designated EOMES in humans), and immature postmitotic neurons, especially neurons of the preplate and early-generated neurons that reside in cortical layer six, express the related Tbr1 gene [Englund et al., 2005; Hevner et al., 2001].
Third, the cerebral cortex receives major contributions from both pallial (dorsal neocortical VZ) and subpallial sources. The latter population consists of neurons that originate in the VZ of the ventral telencephalon, especially the medial ganglionic eminence, and migrate along nonradial pathways through the SVZ and intermediate zone (IZ) to the neocortex, mostly along axonal tracts (see Figure 26-1). These ventral progenitors give rise to inhibitory interneurons that express the neurotransmitter gamma-aminobutyric acid (GABA) [Anderson et al., 1997; Lavdas et al., 1999; Letinic et al., 2002; Wichterle et al., 2001], and constitute 20–30 percent of cortical neurons. This class of neurons modulates cortical output [Cherubini and Conti, 2001; Krimer and Goldman-Rakic, 2001], regulates neuronal proliferation and migration later in corticogenesis [Owens and Kriegstein, 2002], and contributes to postnatal development of cortical circuits [Fagiolini and Hensch, 2000; Huang et al., 1999]. Several distinct subtypes of interneurons have been identified that differ by axonal and dendritic morphology [Lund and Lewis, 1993], chemical markers [Defelipe, 1993; Gonchar and Burkhalter, 1997], connectivity, and physiology [Cauli et al., 1997; Kawaguchi and Kubota, 1996]. The major subtypes of GABAergic interneurons identified by chemical markers include calbindin-, calretinin-, parvalbumin-, somatostatin-, neuropeptide Y- and nitric oxide synthase-positive cells.
Neuronal Migration
Neuroblasts are generated a long distance from their eventual position in the cerebral cortex, a distance that may represent more than a thousand cell body lengths in humans. They undergo a process of movement or migration from either the periventricular zone (glutaminergic neurons) or the ganglionic eminences (GABAergic neurons). Most radial migration occurs by attachment to and locomotion along radial glial fibers, which form an extensive radial lattice that guides radially migrating cells [Gupta et al., 2002; O’Rourke et al., 1992; Rakic, 1971, 1972, 1988]. Radial glia-independent modes of radial migration also occur, particularly by nuclear translocation during the earliest stages while the VZ and cortical plate remain close together [Book and Morest, 1990; Gupta et al., 2002; Morris et al., 1998; Pearlman et al., 1998]. But cells must also receive a polarity signal, adhere to the radial fiber or other extracellular proteins, extend leading processes, translocate their nucleus from the cell body into the leading process to re-establish the cell body in a new location, contract trailing processes, and receive a stop migration signal.
The peak time for neuronal migration is between GW12 and GW20, although migration may continue up to at least GW26 [Evrard et al., 1989; Sidman and Rakic, 1982]. The cortex is formed by an “inside-out” pattern, in which early-generated neurons form the initial cortical plate and later-generated neurons climb past them to progressively more superficial positions. Thus, the earliest neurons to migrate end up in layer six, and the last to migrate in layer two.
Cortical Organization
By approximately 22 weeks, distinctive layers first appear in the cortex. Further maturation consists of additional synaptogenesis, retraction of early axonal collaterals that did not establish appropriate connections, neurotransmitter biosynthesis, and other processes. Most of these processes continue well beyond the perinatal period. The marginal zone of the fetal cortex matures to become layer one, or the molecular layer of mature cortex. The pioneer Cajal–Retzius neurons disappear, leaving a zone of nerve fibers, dendrites, and synapses with only a few cells. The subplate is incorporated into the deep layers of the cerebral cortex. By GW27, all six layers of the mature cortex are visible [Norman et al., 1995]. The cortex also has a columnar or vertical organization that derives from radial (centrifugal) migration of the dominant cell types.
While neuronal identity was specified much earlier in development, migrating neurons only begin the process of differentiation into functional subtypes as their final positions are reached. The critical changes include the appearance of neuronal processes, development of specific neuronal cell membrane properties, and expression of proteins characteristic of a specific type of cell-to-cell transmission. Additional adaptations include further morphologic differentiation, and more complex molecular and physiologic differentiation and synaptic interconnections [Cowan, 1992]. Throughout much of this period, glial cells are dividing and differentiating as well. At least three major types of glial cells are known, including oligodendrocytes, type 1 astrocytes, and type 2 astrocytes. The interactions among these cell types are complex but important in their later role in neuronal migration.
Further Reading
This summary touches on only a few of the advances made over the past decade. Many reviews are available, such as a recent collection of papers in a symposium on “Patterning and Evolving the Vertebrate Forebrain” [Creuzet, 2009; Medina and Abellan, 2009; Merot et al., 2009; Moreno et al., 2009; Saghatelyan, 2009; Subramanian et al., 2009; Suzuki-Hirano and Shimogori, 2009].
Lissencephaly and Subcortical Band Heterotopia
While known from pathological studies for many decades [Culp, 1914; Erhardt, 1914; Matell, 1893], LIS and SBH were brought to modern medical attention between 1963 and 1989 [Barkovich et al., 1989b; Daube and Chou, 1966; Dieker et al., 1969; Miller, 1963]. Their underlying genetic basis first came to light during the same period when several children with LIS and other congenital anomalies associated with deletion of chromosome 17p13.3 were reported, a syndrome now known as Miller–Dieker syndrome [Dobyns et al., 1983; Stratton et al., 1984].
Pathology
LIS is a diffuse brain malformation manifested by a smooth cerebral surface, abnormally thick cortex with four abnormal layers that includes a deep zone of diffuse neuronal heterotopia, and enlarged, dysplastic ventricles [Barkovich et al., 1991; Forman et al., 2005; Norman et al., 1995]. It encompasses the pathologic terms agyria (absent gyri) and pachygyria (broad gyri). SBH consists of symmetric and circumferential bands of gray matter located just beneath the cortex and separated from it by a thin band of white matter. This appearance led to the alternative – although incorrect – term, “double cortex,” for this malformation. The overlying cortex appears normal or mildly simplified as a result of abnormally shallow sulci [Barkovich, 2000; Barkovich et al., 1994, 1989b; Dobyns et al., 1996a].
Several different types of LIS have been recognized, based on pathological features. They are most readily distinguished based on the number of cortical layers, and include four-, three-, and two-layered forms [Forman et al., 2005]. In the common four-layered or classic form (LIS-4L), the gyral malformation can be most severe over either anterior or posterior brain regions. The cortex is 12–20 mm thick and composed of a normal marginal layer, a superficial cellular layer that corresponds to the cortical plate, a cell-sparse zone, and a deep cellular layer composed of heterotopic neurons [Forman et al., 2005]. The two major subtypes can also be distinguished by looking at the transition from the deep cellular layer to the underlying white matter. In the posterior predominant (LIS1 gene) form, the transition is gradual, with no striking features. In the anterior predominant (DCX gene) form, the deep cellular layer transitions to multiple small nodules of subcortical heterotopia, and then to white matter. A few patients with classic LIS have mild to moderate cerebellar hypoplasia.
SBH consists of a normal six-layered cortex, a thin zone of white matter underlying the cortex, and then a zone of dense heterotopic neurons that breaks up into nodules at their lower border, closely resembling the X-linked form of LIS described above [Forman et al., 2005]. LIS-4L and SBH comprise a single malformation spectrum, based on observations of rare patients with areas of LIS that merge into SBH, and of many families with LIS in affected males and SBH in affected females [Des Portes et al., 1997; Dobyns et al., 1996a; Pilz et al., 1999].
The rare two-layered form of lissencephaly (LIS-2L) has been seen only in severe LIS variants with complete agyria and severe brainstem and cerebellar hypoplasia [Forman et al., 2005; Lecourtois et al., 2010]. The molecular layer appears normal, and overlies a single thickened and disorganized cortex with randomly arranged and oriented neurons. The three-layered and two-layered forms of LIS have not been seen with SBH.
The rare three-layered form of LIS (LIS-3L), associated with X-linked lissencephaly with abnormal genitalia (XLAG), consists of a hypercellular marginal or molecular layer, a middle zone with a relative increase in pyramidal neurons, and a deep layer that is relatively thick and composed primarily of small and medium sized neurons [Forman et al., 2005]. Other features include an intermediate-thickness (8–12 mm) cortex, disorganization of the basal ganglia with cysts, gliotic and spongy white matter, and agenesis of the corpus callosum [Bonneau et al., 2002; Forman et al., 2005].
Several rare forms of LIS associated with severe congenital microcephaly have been described, currently designated as microlissencephaly (MLIS), based on birth head circumference smaller than −3 standard deviations at birth. This group may overlap with LIS-2L. The first syndrome with MLIS reported, now known as Norman–Roberts syndrome, consists of severe congenital microcephaly, lissencephaly with diffuse agyria, and several pathological features that differed from classic LIS [Dobyns et al., 1984; Norman et al., 1976]. These included a relatively thin deep cellular layer, no heterotopia of inferior olive or dentate nuclei, and grossly dysplastic cerebellar cortex with reduced granular cells. The authors have seen only one child possibly fitting this syndrome over many years, leading to some doubts regarding its existence. While we believe that this syndrome does exist, several published reports appear to describe different syndromes [Caksen et al., 2004; Iannetti et al., 1993]. In OMIM (number 257320), Norman–Roberts syndrome has been linked to mutations of the RELN gene. This entry is egregiously in error, as no pathological data are available from humans with mutations of RELN or Reelin pathway genes, and individuals with known RELN mutations have normal head size.
Another pathologically distinct three-layered form of LIS (LIS-3L) is associated with microcephalic osteodysplastic primordial dysplasia type 1 (MOPD1). The brain malformation consists of extreme microcephaly, foreshortened frontal lobes, frontal predominant lissencephaly with agyria or pachygyria, agenesis of the corpus callosum, and cerebellar vermis hypoplasia [Juric-Sekhar et al., 2010; Klinge et al., 2002; Meinecke and Passarge, 1991; Ozawa et al., 2005]. The brainstem and cerebellar involvement seems less severe than lissencephaly with cerebellar hypoplasia (LCH). The dysplastic cortex is characterized by extensive but thin glioneuronal heterotopia, a thin superficial layer that appears hyperconvoluted in places (thus somewhat resembling polymicrogyria), a thin second layer of disorganized medium to large neurons, and a thick third layer consisting of mostly small neurons [Juric-Sekhar et al., 2010]. The glioneuronal heterotopia, lack of white-matter cystic changes, the brainstem and cerebellar hypoplasia, and other features distinguish this from the XLAG-type of LIS-3L.
The most severe form of true lissencephaly may be the form reported as “familial lissencephaly with extreme neopallial hypoplasia” that we now designate as Barth-type microlissencephaly [Barth et al., 1982; Dobyns and Barkovich, 1999; Kroon et al., 1996]. The pathological features include extreme congenital microcephaly, severe LIS with diffuse agyria, disorganized cortex with four layers resembling classic LIS-4L, except for a relatively thin deep cellular layer, small collections of cells with scant cytoplasm (possible germinal cells) in the leptomeninges, absent olfactory nerves, severe optic nerve hypoplasia, small thalami resembling streaks, agenesis of the corpus callosum and other commissures, severe brainstem hypoplasia with absent cerebral peduncles, transverse pontine fibers, inferior olives and pyramids, and extreme cerebellar hypoplasia with absent folia.
Finally, use of the old terms “type 1” and “type 2” LIS is strongly discouraged in favor of the new descriptive terms used above. The old “type 1” was first used to refer to the four-layered form, but was later applied to anything that was not “type 2.” The old “type 2” form is now designated as cobblestone malformation, discussed in the next section. Only the most severely affected patients have agyria or pachygyria. A few reports have used the term “type 3” LIS to refer to a rare condition with severe fetal akinesia, microcephaly, and undersulcated brain surface [Allias et al., 2004; Encha Razavi et al., 1996]. The cortex in this form appears thin, rather than thick, and has evidence of neuronal cell loss, suggesting a prenatal-onset degenerative disorder that would best not be classified as a form of LIS. In addition, both severe congenital microcephaly and more severe forms of polymicrogyria are commonly misidentified as LIS.
Brain Imaging
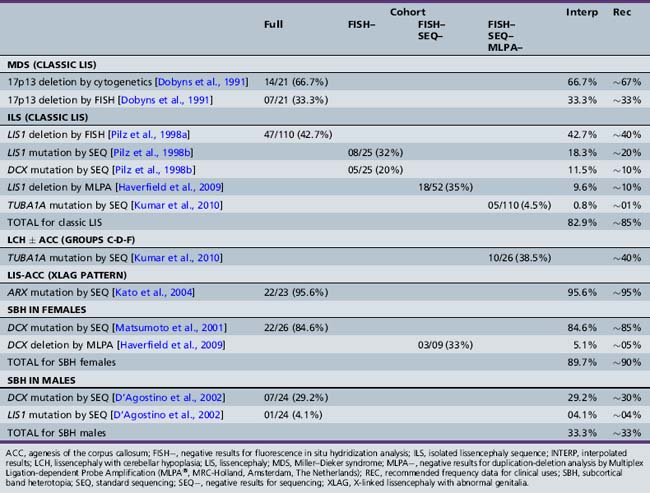
Most of the distinguishing features seen on pathological examination can be viewed by brain imaging as well (Figure 26-2). The different types of LIS and SBH may be distinguished by both the pattern and the severity of the malformation. Recognition of these patterns has become essential for syndrome and molecular diagnosis, and for assessing prognosis and genetic risk [Dobyns et al., 1999b; Kato and Dobyns, 2003; Pilz et al., 1998b]. The different patterns of LIS and SBH associated with known syndromes and causal genes are summarized in Table 26-1.
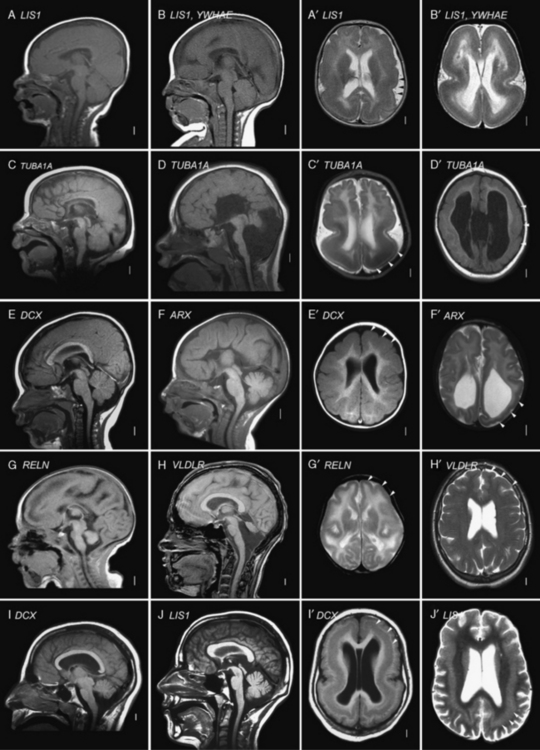
Fig. 26-2 Subtypes of lissencephaly.
(A–H, Reprinted from Dobyns WB. The clinical patterns and molecular genetics of lissencephaly and subcortical band heterotopia. Epilepsia 51[Suppl. 1]:5–9, 2010, with permission from Wiley–Blackwell. I, Courtesy of Dr. William B Dobyns, University of Washington, Departments of Pediatrics and Neurology, Seattle, Washington. These images were selected from patients LR08-316 [A–A′], LR08-401 [B–B′], LR07-008 [C–C′], LR07-244 [D–D′], LR06-020 [E–E′], LR04-245 [F–F′], LP95-137a2 [G–G′], LR08-330 [H–H′], LP94-049 [I–I′], and LP94-051 [J–J′].)
Table 26-1 Lissencephaly Syndromes, Inheritance and Genes
| Lissencephaly Type and Syndrome | Inheritance | Genes |
|---|---|---|
| LIS-4L SEVERE WITH NO GRADIENT | ||
| Miller–Dieker syndrome (deletion 17p13.3) | SpAD | LIS1-YWHAE |
| Isolated lissencephaly sequence, severe X-linked type | XL | DCX |
| Barth-type microlissencephaly | AR | Unknown |
| LIS-4L POSTERIOR > ANTERIOR GRADIENT AND VARIANTS | ||
| Isolated lissencephaly sequence with p > a gradient | SpAD | LIS1, TUBA1A |
| Isolated lissencephaly sequence with p > a gradient | AR | Unknown |
| Subcortical band heterotopia with p > a gradient | SpAD | LIS1 |
| Subcortical band heterotopia with p > a gradient | AD | Unknown |
| Lissencephaly with cerebellar hypoplasia with p > a gradient plus mild cerebellar (vermis) hypoplasia | SpAD | LIS1 |
| Lissencephaly with cerebellar hypoplasia with central > posterior > anterior gradient (tubulin-type), moderate cerebellar hypoplasia, and often ACC (group f with central gradient) | SpAD | TUBA1A |
| LIS-4L ANTERIOR > POSTERIOR GRADIENT AND VARIANTS | ||
| Isolated lissencephaly sequence with a > p gradient | XL | DCX |
| Isolated lissencephaly sequence with a > p gradient | AR | Unknown |
| Subcortical band heterotopia with a > p gradient | XL | DCX |
| Baraitser–Winter syndrome | SpAD | Unknown |
| LIS-2L SEVERE WITH NO GRADIENT | ||
| Lissencephaly with cerebellar hypoplasia (tubulin-type) with severe cerebellar hypoplasia and ACC (groups c, d, and f with no gradient) | SpAD | TUBA1A |
| LIS-3L POSTERIOR > ANTERIOR GRADIENT VARIANT | ||
| X-linked lissencephaly with abnormal genitalia (XLAG) | XL | ARX |
| XLAG-like syndrome with microphthalmia, and cleft lip and palate | AR? | Unknown |
| LIS-3L ANTERIOR > POSTERIOR GRADIENT VARIANT | ||
| Microlissencephaly MOPD1-type with cerebellar hypoplasia | AR | Unknown |
| LIS-RELN ANTERIOR > POSTERIOR GRADIENT | ||
| LCH RELN-type with severe hippocampal and cerebellar hypoplasia (group b, frontal predominance) | AR | RELN, VLDLR |
ACC, agenesis of the corpus callosum; AD, autosomal-dominant; AR, autosomal-recessive; LCH, lissencephaly with cerebellar hypoplasia; MOPD, microcephalic osteodysplastic primordial dysplasia; SpAD, sporadic with presumed autosomal-dominant inheritance caused by new mutations; XL, X-linked.
For all forms of true LIS, the brain surface appears smooth with areas of absent gyri (agyria) and abnormally wide gyri (pachygyria), and abnormally thick cerebral cortex [Barkovich et al., 1991; Dobyns and Truwit, 1995]. In normal brains, most gyri are approximately 1–1.5 cm wide and the normal cortex 3–4 mm thick, but thicker in the primary motor cortex and thinner in the primary visual cortex. In all types of LIS, gyri are typically 3 cm wide or more, and the cortex 8–20 mm thick, although this varies with the type (see Figure 26-2A–G, with Figure 26-2H being an exception). Several distinct types of LIS are associated with agenesis of the corpus callosum, moderate to severe cerebellar hypoplasia, designated LCH, or both [Ross et al., 2001].
In SBH (see Figure 26-2I–J), the brain surface appears superficially normal, except that the sulci or crevices between gyri tend to be very shallow (less than 1 cm instead of 1–3 cm), and the cortex is normal and not thick [Barkovich et al., 1994; Dobyns et al., 1996a]. Just beneath the cortex, however, often separated from it by just a few millimeters of white matter, lies a smooth band of neurons that never reached the true cortex. The inner margin of the band is usually smooth, while the outer margin may appear smooth (with thick bands) or follow interdigitations of the overlying cortex (with thin bands).
The most common or classic form of LIS seen on brain imaging corresponds to LIS-4L, and occurs in a continuous series with SBH. The severity varies from complete or nearly complete agyria (grades 1 and 2, see Figure 26-2B); to mixed agyria-pachygyria (grade 3, see Figure 26-2A and C); and then to extensive pachygyria (grade 4, see Figure 26-2D and E); mixed pachygyria and SBH (grade 5, not shown); and finally, SBH alone (grade 6, see Figure 26-2I and J). Both LIS-4L and SBH may be seen with either a frontal or posterior predominant distribution. Thus, the gradient of LIS and SBH can be anterior more severe than posterior (a > p), posterior more severe than anterior (p > a), or anterior equal to posterior (a = p). In LIS-4L, the cortex is usually 12–20 mm thick. Common associated malformations include thick and rounded hippocampi, enlarged posterior portions of the lateral ventricles, and flat anterior portion of the corpus callosum.
Several other types of LCH may be differentiated, based on brain imaging. A few patients with classic LIS have moderate vermis-predominant cerebellar hypoplasia, which corresponds to the previous LCH group a [Ross et al., 2001]. The next consists of intermediate severity LIS with mixed agyria and pachygyria that appears most severe in central (posterior frontal and anterior parietal) regions (see Figure 26-2D). Subtle asymmetry is often seen, as well as hypogenesis or agenesis of the corpus callosum. Another more severe variant of LCH consists of diffuse agyria with a cortex that appears only moderately thick (not shown). The lower margin of the cortex may be smooth or follow an undulating course. Associated abnormalities include severe hypoplasia of the hippocampus, variable agenesis of the corpus callosum, and severe brainstem and cerebellar hypoplasia. This severe variant corresponds to LIS-2L, and combines the prior LCH groups c, d, and f [Forman et al., 2005; Kumar et al., 2010; Ross et al., 2001].
A less severe LCH variant, which was previously designated as LCH group b [Ross et al., 2001], consists of frontal-predominant lissencephaly consisting of mild to moderate pachygyria, mildly thick 8–10 mm cortex, very small globular hippocampi, mildly small brainstem, and diffuse severe hypoplasia of the cerebellum with absent or nearly absent folia (see Figure 26-2G and H). This pattern has been associated with mutations of Reelin pathway genes.
The LIS variant seen in children with the XLAG syndrome corresponds to LIS-3L [Bonneau et al., 2002; Forman et al., 2005]. It is characterized by mixed agyria and pachygyria that appears most severe over the temporal lobes, next most severe over the parietal and occipital lobes, and least severe over the frontal lobes; the cortex is only moderately thick, usually 8–10 mm (see Figure 26-2F). Associated abnormalities include agenesis of the corpus callosum that is most often complete, abnormal basal ganglia with indistinct borders and cysts, and diffuse abnormal (high T2, low T1) signal of white matter. The brainstem and cerebellum appear normal.
Clinical Features
A few children feed poorly from the first weeks of life, but this often improves unless they have one of the severe LIS variants. Most feed reasonably well for the first several years, except that many have difficulty during intercurrent illnesses. Feeding often worsens later, especially after about 3 years, with increased aspiration, decreased feeding tolerance, and recurrent pneumonia. These problems are frequently related to worsening epilepsy and gastroesophageal reflux, whether obviously symptomatic or not. They lead to placement of gastrostomy tubes and operations to reduce reflux (fundal plications) in many affected children, although the age varies widely within the first decade [Dobyns et al., 1992].
Epilepsy
Most – indeed, probably all – children with LIS have seizures. The onset is usually between 3 and 12 months, but may be later. Between 35 and 85 percent of children with classic LIS develop infantile spasms in the first year of life, although hypsarrhythmia is usually absent. After 1 year, they typically have continued mixed seizure types, including epileptic spasms, typically presenting on awakening, myoclonic, tonic, and tonic-clonic seizures. Many meet criteria for Lennox–Gastaut syndrome, which can be associated with a decline in skills with poor seizure control. In general, the same treatment strategies used for Lennox–Gastaut syndrome generally may be used in patients with LIS or SBH. In children with XLAG, epilepsy is nearly continuous. In both classic LIS and XLAG, studies in mouse mutants have shown deficiencies in cortical interneurons that use GABA as their primary neurotransmitter [Marsh et al., 2009; McManus et al., 2004]. Thus, GABAergic medications have some theoretical basis for use in these children.
Prognosis
In contrast, most patients with SBH and rare patients with partial forms of LIS have mild to moderate mental retardation, although both normal intelligence and severe mental retardation have been seen. Other clinical features are minimal pyramidal signs and dysarthria [Barkovich et al., 1994; D’Agostino et al., 2002; Dobyns et al., 1996a]. Seizures usually begin in childhood but may appear much later, and multiple types occur that may be difficult to control. Cognitive development may slow after onset of seizures. The frequency and severity vary greatly. EEG investigations usually demonstrate generalized spike-and-wave discharges or multifocal abnormalities [Battaglia, 1996; D’Agostino et al., 2002; Palmini et al., 1991]. Neurologic outcome usually correlates with the thickness of the subcortical band heterotopia and simplification of the gyral pattern, as seen on magnetic resonance imaging (MRI).
Syndromes, Genetics, and Molecular Basis
While older reports suggested the possibility of extrinsic (nongenetic) causes of LIS, such as prenatal cytomegalovirus exposure or perfusion failure [Dobyns et al., 1992; Norman et al., 1976], clinical experience suggests that all or almost all LIS is genetic. Mutations of the six known LIS genes, including ARX, DCX, LIS1, RELN, TUBA1A, and VLDLR, account for more than 80 percent of patients [Boycott et al., 2005; Gleeson et al., 1998; Hong et al., 2000; Keays et al., 2007; Kitamura et al., 2002; Reiner et al., 1993]. However, several of these are associated with one or more specific LIS or SBH syndromes with different presentations. The contributions of these six genes and the specific mutational mechanisms have been derived from separate studies over many years, making comparisons difficult. An update of the prior results in light of current knowledge is shown in Table 26-2 [Dobyns, 2010]. A review of the major LIS syndromes and their molecular mechanisms is presented in the following sections.
Miller–Dieker Syndrome
Miller–Dieker syndrome is a striking multiple congenital anomaly syndrome, characterized by classic LIS (LIS-4L), typical facial appearance, and variable other birth defects, such as heart malformations. The facial features include prominent forehead, bitemporal hollowing, short nose with upturned nares, protuberant upper lip with thin vermilion border, and small jaw. The brain malformation consists of severe LIS with no apparent gradient (see Figure 26-2B), although rare patients may have the same posterior more severe than anterior gradient seen in patients with isolated lissencephaly sequence with LIS1 mutations [Cardoso et al., 2003; Dobyns et al., 1991]. All patients with Miller–Dieker syndrome have large deletions of chromosomal region 17p13.3 that include LIS1, YWHAE, and all intervening genes, which indicates that the YWHAE gene is an important modifying factor for lissencephaly. Several genes in this region are known oncogenes, and at least two patients with Miller–Dieker syndrome have had childhood cancers [Czuchlewski et al., 2008; Ueda et al., 2006].
Isolated Lissencephaly Sequence
Isolated lissencephaly sequence, which is by far the most common of the LIS syndromes, consists of classic LIS (LIS-4L) with a normal facial appearance, except for mild bitemporal hollowing and small jaw [Dobyns et al., 1992, 1984]. Different patterns of LIS have been found with mutations of the three known causative genes – DCX, LIS1, and TUBA1A. A few patients with mutations of these genes – especially TUBA1A – also have cerebellar hypoplasia. Isolated lissencephaly sequence associated with mutations of the X-linked DCX gene is characterized by either severe lissencephaly with no clear gradient, or lissencephaly with an anterior more severe than posterior gradient (see Figure 26-2E), and normal facial appearance [Dobyns et al., 1999b; Pilz et al., 1998b]. Isolated lissencephaly sequence associated with mutations or deletions of the LIS1 gene or mutations at codon 402 in TUBA1A (p.R402C or p.R402H) is characterized by lissencephaly with a posterior more severe than anterior gradient or, less often, an anterior equal to posterior gradient (see Figure 26-2A and C). Facial appearance may be normal, or have subtle dysmorphism similar to that in Miller–Dieker syndrome, but much less severe [Cardoso et al., 2003; Dobyns et al., 1992; Kumar et al., 2010].
For isolated lissencephaly sequence, the parents should be tested whenever mutations of LIS1 or TUBA1A have been found. When the results are normal, the recurrence risk is low, as evidenced by lack of recurrence in more than 100 such families seen. However, the possibility of recurrence exists due to mosaicism in one parent, so that the availability of prenatal diagnosis should be discussed for any future pregnancies. When mutations of DCX are found, the rules for X-linked inheritance apply [Dobyns et al., 2004]. Carrier testing in mothers is important, as many are found to be carriers [Gleeson et al., 2000a; Matsumoto et al., 2001], especially when the proband has a less severe phenotype [Guerrini et al., 2003]. The recurrence risk is 50 percent when the mother is found to be a carrier [Matsumoto et al., 2001]. Testing in fathers of female probands should be considered as well, as one father has been found who is a mosaic carrier of the mutation found in his affected daughter. Recurrence in siblings has been reported in families when results of mutation analysis of the proband are negative [Gleeson et al., 2000b; Kuzniecky, 1994; Ramirez et al., 2004], so counseling for a 10–15 percent recurrence risk is suggested in this situation. When no testing has been done, counseling is difficult.
Subcortical Band Heterotopia
SBH is only rarely associated with other congenital anomalies. Most patients are female because the most common cause is heterozygous mutations of the DCX gene on the X chromosome [Dobyns, 2010; Dobyns et al., 1996a; Gleeson et al., 2000a; Matsumoto et al., 2001]. This represents the heterozygous or carrier phenotype found in female relatives of males with DCX-related lissencephaly. Many affected males, however, also have been reported [D’Agostino et al., 2002]. Mutations of DCX and LIS1, the same two genes that cause isolated lissencephaly sequence, have been identified that result in somewhat different patterns of malformation. SBH associated with mutations of DCX gene is characterized by diffuse thick bands with no apparent gradient, or by partial frontal thin bands [Gleeson et al., 2000a; Matsumoto et al., 2001]. SBH associated with mutations or deletions of the LIS1 gene is characterized by partial posterior thin or intermediate bands with an obvious posterior more severe than anterior gradient. This is a rare cause of SBH, and some patients have mosaic mutations of LIS1 [D’Agostino et al., 2002; Mineyko et al. 2010; Pilz et al., 1999; Sicca et al., 2003] or deletions of 17p13.3 (unpublished data). Several families have been reported with posterior SBH, familial recurrence, and negative testing for both DCX and LIS1 [Deconinck et al., 2003].
Baraitser–Winter Syndrome
The rare Baraitser–Winter syndrome (BWS) consists of facial dysmorphism, typically including trigonocephaly and shallow orbits, colobomas of both iris and retina, and lissencephaly (LIS-4L) with an anterior more severe than posterior gradient [Baraitser and Winter, 1988; Forman et al., 2005; Ramer et al., 1995; Rossi et al., 2003; Verloes, 1993]. The lissencephaly usually is relatively mild, with pachygyria most severe in the midfrontal region that undergoes transition posteriorly to subcortical band heterotopia and then to a more normal gyral pattern, thus resembling the less severe end of the DCX spectrum. Familial recurrence has never been reported, which suggests new-mutation autosomal-dominant inheritance.
LIS Variants
At least seven further types of LIS have been described in rare patients with variant types of LIS, based on neuropathology, brain imaging, or both (see Table 26-1). The key differentiating features in the brain include severe congenital microcephaly (in 2 out of 7), variant cortical histology (2L in 1 of 7 and 3L in 2 of 7), agenesis of the corpus callosum (in 5 of 7), and variable but often severe cerebellar hypoplasia (in 6 of 7). The combination of LIS with severe congenital microcephaly is designated as microlissencephaly (MLIS) only when the cortex is abnormally thick; otherwise, it is classified as “microcephaly with a simplified gyral pattern.” The previous classification of LCH into six groups is updated to include several other types [Ross et al., 2001].
Lissencephaly with Cerebellar Hypoplasia RELN-Type
This rare syndrome was first reported as a variant of the so-called dysequilibrium syndrome, and later as “neuronal migration defect, cerebellar hypoplasia, and lymphedema” [Hourihane et al., 1993; Schurig et al., 1981]. The brain malformation consists of moderate frontal-predominant LIS (pachygyria), only moderately thick cortex (8–10 mm), small globular hippocampus, and small, often afoliar, cerebellum, a pattern previously classified as LCH group b (see Figure 26-2G and H) [Ross et al., 2001]. No data on the neuropathology in humans are available, although mouse models have a classic “inverted” cortex [D’Arcangelo et al., 1995; Falconer, 1951]. The phenotype is characterized by moderate to profound mental retardation, delayed ambulation, nonprogressive truncal and peripheral ataxia, and occasional seizures, in addition to the brain malformation [Glass et al., 2005]. Mutations of either RELN or VLDLR have been found in several patients in this group [Boycott et al., 2009, 2005; Chang et al., 2007; Hong et al., 2000; Ozcelik et al., 2008; Zaki et al., 2007].
Lissencephaly with Cerebellar Hypoplasia Classic-Type
The next, and possibly most common, type resembles isolated lissencephaly sequence (ILS), with the addition of mild cerebellar vermis hypoplasia, which was previously designated as LCH group a [Ross et al., 2001]. The LIS may have either frontal or posterior predominance, and the phenotype closely resembles ILS. As in ILS, the frontal-predominant group is associated with mutations of DCX, and the posterior-predominant group with mutations of LIS1 or TUBA1A. Some patients have negative testing for all three genes. The pathology probably consists of LIS-4L.
Lissencephaly with Cerebellar Hypoplasia Tubulin-Type 1
Another phenotype consists of moderate but variable LIS (pachygyria) with central (posterior frontal and anterior parietal) predominance, variable hypogenesis of the corpus callosum, and moderate cerebellar hypoplasia, a phenotype that was previously included in LCH group f (see Figure 26-2D) [Ross et al., 2001]. This subgroup was delineated in a subgroup of children with mutations of TUBA1A [Kumar et al., 2010; Morris-Rosendahl et al., 2008; Poirier et al., 2007]. The phenotype is usually severe, similar to isolated LIS, and the pathology probably varies from LIS-4L to more severe changes, based on pathological data from fetal cases [Fallet-Bianco et al., 2008].
Lissencephaly with Cerebellar Hypoplasia Tubulin-Type 2
A more severe but related phenotype consists of severe LIS with diffuse agyria, agenesis or severe hypogenesis of the corpus callosum, and severe diffuse brainstem and cerebellar hypoplasia, a group that had previously been separated into LCH groups c, d, and f [Kumar et al., 2010; Ross et al., 2001]. The phenotype is very severe, with most patients surviving only a few weeks to months. The neuropathology corresponds to the rare two-layered form of LIS [Forman et al., 2005; Lecourtois et al., 2010]. Several have had mutations of TUBA1A [Kumar et al., 2010].
Microlissencephaly Microcephalic Osteodysplastic Primordial Dwarfism 1-Type
MLIS occurs in some patients with MOPD1, a syndrome that is difficult to distinguish from severe forms of Seckel’s syndrome [Juric-Sekhar et al., 2010; Klinge et al., 2002; Meinecke and Passarge, 1991; Ozawa et al., 2005]. The phenotype consists of severe prenatal growth deficiency and microcephaly, sparse hair and dry scaling skin, skeletal anomalies such as platyspondyly, slender ribs, short and bowed proximal humeri and femurs, small iliac wings, dysplastic acetabulum and small hands and feet, and profound developmental handicaps. A few have developed aplastic anemia, another overlap with Seckel’s syndrome. The neuropathology consists of a variant form of LIS-3L with frontal predominance.
Microlissencephaly Barth-Type
The Barth-type of MLIS is possibly the most severe of all the known LIS syndromes. The phenotype consists of polyhydramnios, probably due to poor fetal swallowing, severe congenital microcephaly (birth OFC approximately 28 cm), weak respiratory effort, and survival for only a few hours or days [Barth et al., 1982; Dobyns and Barkovich, 1999; Kroon et al., 1996]. The neuropathology consists of a variant form of LIS-4L with extreme hypoplasia of many structures, as described above.
X-linked Lissencephaly with Abnormal Genitalia
Children with XLAG are almost all males and have severe clinical problems [Berry-Kravis and Israel, 1994; Bonneau et al., 2002; Dobyns et al., 1999a; Kato et al., 2004; Uyanik et al., 2003]. Frequent, and sometimes continuous, seizures are typically seen on the first day of life, often in the delivery room. Other problems include poor temperature regulation leading to persistent hypothermia, hypotonia, very poor feeding, chronic secretory diarrhea, and ambiguous or severely hypoplastic genitalia. The seizures generally do not respond well to therapy, and are accompanied by an infancy-onset dyskinesia that may be difficult to distinguish from seizures. The feeding problems and diarrhea combine to lead to very poor nutrition. Some female relatives, especially sisters and maternal aunts (non-mothers), have isolated agenesis of the corpus callosum [Marsh et al., 2009; Proud et al., 1992]. Mutations of the ARX gene have been found in almost all patients [Kato et al., 2004; Kitamura et al., 2002] with this syndrome. The XLAG-associated form of LIS-3L is recognizable on brain imaging, and strongly points to this diagnosis (see Figure 26-2F). However, the same pattern has been seen in two children with a separate syndrome that includes microphthalmia with cleft lip and palate.
Notably, less severe mutations of ARX have been found in boys with cryptogenic infantile spasms, infancy-onset dyskinesia, and some less specific mental retardation and epilepsy syndromes [Bienvenu et al., 2002; Kato et al., 2003, 2007; Partington et al., 2004; Scheffer et al., 2002; Stromme et al., 2003; Stromme et al., 2002a, 2002b; Turner et al., 2002]. Studies in a mouse model have found that nonradial migration of inhibitory interneurons from the embryonic ganglionic eminence (the origin of the basal ganglia) to the neocortex is disrupted with loss of Arx [Kitamura et al., 2002; Marsh et al., 2009].
Genetic Counseling
All forms of LIS and SBH are genetic, and genetic testing is currently available for more than 80 percent of patients, although this varies with the phenotype (see Table 26-2) [Dobyns, 2010; Haverfield et al., 2009]. When LIS or SBH is suspected but the exact syndrome diagnosis is uncertain, the most productive order of testing begins with analysis for deletions of chromosome 17p13.3 that include the LIS1 gene, using chromosome microarray or other methods, followed by sequencing of LIS1, DCX, and TUBA1A, and sometimes ARX if the phenotype is compatible. However, details of the phenotype can be used by experienced clinicians to change the order of tests, such as testing DCX first in females with SBH, or ARX first for males with XLAG. Genetic testing for LIS and SBH is important, as several syndromes are associated with high risks of recurrence. This risk is especially great for parents who are carriers of a rearrangement of chromosome 17, and for mothers who carry ARX or DCX mutations, as both are X-linked. When the causal gene is unknown or unavailable for testing, empiric recurrence risks based on the known or most likely pattern of inheritance are appropriate. These are listed in Table 26-1.
Cobblestone Malformations
Pathology
The brain malformations seen in WWS, MEB, or FCMD consist of poor gyral development, cerebral and cerebellar cortical dysplasia, hydrocephalus, brainstem hypoplasia with dysplasia of the inferior olives and dentate nuclei, and hypoplasia of the corticospinal tracts in brainstem and spinal cord [Bordarier et al., 1984; Dobyns et al., 1985; Towfighi et al., 1984; Williams et al., 1984]. Studies of affected human fetuses demonstrate a striking cerebral and cerebellar cortical dysplasia with discontinuities in the basal lamina at the pial surface (glia limitans) that allow abnormal migration of neurons and glia beyond the cortical plate and into the leptomeninges [Miller et al., 1991; Squier, 1993; Takada et al., 1987]. The same changes are seen in several animal models [Moore et al., 2002; Willer et al., 2004]. The cerebellar cortical cysts result from entrapment of islands of meningeal tissue between adjacent folia [Muntoni et al., 2009].
The postnatal cortical malformation consists of an undersulcated and subtly pebbled surface that includes areas resembling agyria, pachygyria, or polymicrogyria, although the histological appearance is very different. The pebbled surface appearance prompted the name “cobblestone cortex,” first proposed by Haltia [Dubowitz, 1996]. The cortex is thick and dysplastic, with no recognizable layers. The original glia limitans is found in the middle of the cortex, which is disrupted by abnormal vascular channels and fibroglial bands throughout that extend into and often obstruct the subarachnoid space. The cortical malformation is most severe with a smooth surface in WWS, and progressively less severe in the other syndromes in this probably continuous series [Dobyns et al., 1985, 1989; Dubowitz, 1994, 1996; Haltia et al., 1997; Takada et al., 1988; Walker, 1942]. The white matter is poorly myelinated, with numerous heterotopic neurons and microscopic cysts. The cerebellum is small and dysplastic, with cerebellar cortical cysts. Other brain abnormalities include obstructive hydrocephalus, brainstem hypoplasia with dysplasia of the inferior olives and dentate nuclei, and hypoplasia of the corticospinal tracts in brainstem and spinal cord. In a few patients, absent septum pellucidum, absent corpus callosum, or occipital cephaloceles may be noted. The brain malformation is frequently associated with eye malformations. Skeletal muscle biopsies show changes of CMD and hypoglycosylated α-dystroglycan [Brockington and Muntoni, 2005; Mercuri et al., 2006; Muntoni et al., 2002].
Only limited human pathological data are available for the cobblestone malformations that are not associated with CMD. Analysis of a Gpr56 knockout mouse shows discontinuities in the pial surface basal lamina and migration of neurons and glia into the leptomeninges, just as in the CMD-associated cobblestone malformations [Li et al., 2008]. The first neuropathology data in humans were recently reported in abstract form, and confirm the cobblestone-type cortical malformation with widespread glioneuronal heterotopia [Fallet-Bianco et al., 2010].
Brain Imaging
The brain imaging changes of CMD-associated cobblestone malformation present a continuous series of malformations that begins with the severe changes of WWS and ends with normal brain imaging. However, most patients cluster into one of the defined syndromes, which consist of WWS, an intermediate group classified as MEB, and progressively less severe forms classified as FCMD, CMD with cerebellar dysplasia and cysts or CMD with microcephaly, and CMD with normal brain imaging. The key imaging features are most easily recognized for WWS, and are collectively pathognomonic for this disorder [Clement et al., 2008; Dobyns et al., 1989; Jissendi-Tchofo et al., 2009; Mercuri et al., 2006; van der Knaap et al., 1997].
The imaging abnormalities seen in WWS begin with macrocephaly and prominent forehead, resulting from existing or prior hydrocephalus, and reduced size and partial obliteration of extra-axial spaces, which is especially prominent between the cerebral hemispheres (Figure 26-3C). The cerebral surface is undersulcated, usually with diffuse agyria. The cerebral cortex is moderately thick, usually about 7–10 mm unless thinned by hydrocephalus. The cortical–white matter border is jagged, with frequent vertical (perpendicular to the cortical–white matter border) striations, which differs from the chaotic striations typical of true polymicrogyria. Just beneath the cortex, streaks of laminar subcortical heterotopia are seen that differ from typical subcortical band heterotopia based on their beaded and discontinuous appearance. The white matter has very abnormal signal (bright on T2 and dark on T1 MRI sequences; dark on computed tomography [CT] scan) and may have small cysts. The white-matter volume may be normal, or thinned by hydrocephalus. The third and lateral ventricles are enlarged and may be very large and rounded, reflecting active hydrocephalus. Very rarely, the ventricles may be small. The corpus callosum is present, although frequently thin.
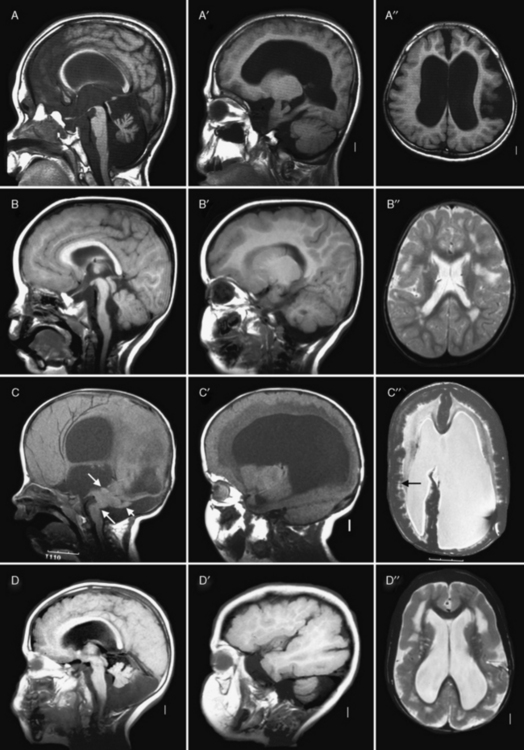
Fig. 26-3 Subtypes of cobblestone cortical malformations.
Midsagittal (left column), parasagittal (middle column), and axial (right column) magnetic resonance images from patients with four different cobblestone malformation syndromes. A–A″, The top row demonstrates brain abnormalities in Fukuyama congenital muscular dystrophy. These images show mild frontal pachygyria with 7- to 8-mm-thick cortex, normal to mildly simplified gyral pattern posteriorly, widely open sylvian fissures, especially on the left, moderately enlarged lateral ventricles, intact but stretched corpus callosum, normal brainstem, and moderate cerebellar hypoplasia and atrophy. These images are from a Japanese child with Fukuyama’s congenital muscular dystrophy (FCMD), who most likely is homozygous for the Japanese founder mutation consisting of addition of a 3-kb retrotransposon into the 3′ untranslated region of the FCMD gene [Kobayashi et al., 1998]. B–B″, The second row shows changes in muscle-eye-brain disease. These images show moderate frontal pachygyria with an 8- to 12-mm-thick cortex that extends into the parietal lobe, patches of abnormal white matter with high T2 signal intensity, hypoplastic brainstem, especially in the pons, moderate cerebellar hypoplasia, and a few small cysts in the high white matter. The patient was a boy with a homozygous mutation of POMGnT1 in exon 20: c.1813delC and H573fs (patient CC described by Yoshida and colleagues [Yoshida et al., 2001] and Taniguchi and associates [Taniguchi et al., 2003]). C–C″, The third row shows changes in severe Walker–Warburg syndrome. These images show diffuse agyria with an irregular surface, beaded subcortical heterotopia (black arrow in C″), diffuse abnormal signal of white matter with very high T2 signal intensity, very thin corpus callosum, and moderate to severely enlarged lateral ventricles. Posterior fossa abnormalities include severe hypoplasia of the brainstem, enlarged tectum (top white arrow in C), classic Walker–Warburg kinking at the junction of the pons and midbrain (bottom long white arrow in C), severe cerebellar hypoplasia (bottom short white arrow in C), small fluid collection in the posterior fossa, and overall small posterior fossa. No mutation has been identified, although only POMT1 has been tested. D–D″, The bottom row shows the changes in bilateral frontoparietal polymicrogyria, with extensive involvement of the frontal and parietal lobes, extended sylvian fissure, patches of very bright T2 signal of white matter (again, more severe frontally), mildly enlarged lateral ventricles, thin corpus callosum, and hypoplasia of the brainstem and cerebellum. The patient was a girl with a missense mutation of the GPR56 gene in exon 3: 112C3T and R38W [Piao et al., 2004].
(Courtesy of Dr. William B Dobyns, University of Washington, Departments of Pediatrics and Neurology, Seattle, Washington. These images were selected from patients LR01-058 [A], LP95-146 [B], LR00-181 [C], and LP93-017 [D]).
The brainstem and cerebellum in WWS (see Figure 26-3C) are remarkably dysplastic, with a kink at the midbrain–pons junction in which the midbrain is angled dorsally with respect to the pons [Jissendi-Tchofo et al., 2009]. The lower brainstem appears small, with moderate to severe hypoplasia of the medulla and pons, near-absence of the basis points, and ventral midline clefts of the ventral pons. But the midbrain and especially the tectum are abnormally large. The cerebellum is small, with the vermis more severely involved than the hemispheres, a dysplastic foliar pattern, and often small cysts within or near the cerebellar cortex. The posterior fossa may be enlarged and a few patients have small occipital meningoceles.
The imaging abnormalities in MEB are similar but consistently less severe than in WWS (see Figure 26-3B). The macrocephaly and hydrocephalus are common in MEB as well, while partial obliteration of extra-axial spaces is less extensive. The cerebral surface is again undersulcated, but with frontal-predominant pachygyria rather than agyria; posterior-predominant pachygyria occurs but appears to be rare. Some areas resembling polymicrogyria may be seen. The jagged cortical–white matter border and vertical striations are similar, while fewer streaks of subcortical heterotopia are seen. The white matter has very abnormal signal, similar to WWS in infants, but over time this evolves from diffuse to patchy to minimal signal changes. The frequency of hydrocephalus and thinning of the white matter and corpus callosum is probably similar.
The brainstem and cerebellum in MEB (see Figure 26-3B) are also dysplastic but less severely so than in WWS. The brainstem lacks the kink, but the lower brainstem, especially the pons, appears small while the midbrain and tectum are enlarged. The cerebellar changes are similar but, on average, less severe than in WWS, except that cerebellar cortical cysts may be more common. This may be due to a high frequency of cerebellar cysts in MEB patients with mutations of the POMGnT1 gene [Muntoni et al., 2009].
The imaging abnormalities in FCMD (see Figure 26-3A) are similar to MEB but less severe. Hydrocephalus is uncommon. The cortical malformation is less severe but still frontal-predominant, although some patients have severe temporal lobe agyria. The brainstem and cerebellum most often appear normal. In contrast, patients with mental retardation and cerebellar cysts have relatively normal forebrain structures and cerebral cortex, although review of some published images shows that the cortex is mildly dysplastic. The cerebellum mimics the appearance seen in MEB.
The brain imaging abnormalities in the cobblestone-like disorders without CMD (see Figure 26-3D) are similar to those in MEB, including the frontal-predominant pachygyria, moderately thick cortex with vertical striations, patchy white-matter abnormalities, hydrocephalus, and brainstem- and vermis-predominant cerebellar hypoplasia and dysplasia [Chang et al., 2003; Van Maldergem et al., 2008]. The brainstem is usually not as thin as is seen in MEB.
Clinical Features
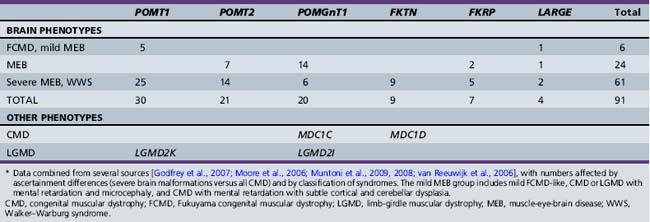
In all of the cobblestone malformation syndromes associated with CMD, the phenotype consists of moderate to profound mental retardation, severe hypotonia, mild distal spasticity, and poor vision. WWS is the most severe, FCMD and mental retardation with cerebellar cysts the least severe, and MEB in the middle. This is in part a semantic issue, as MEB is the diagnosis used for any intermediate phenotype. Several less severe CMD-associated phenotypes without brain malformations have been associated with different mutations of the same genes. The other (non-CMD) cobblestone syndromes have brain phenotypes that resemble FCMD or MEB, but without the eye or muscle manifestations [Clement et al., 2008; Godfrey et al., 2007; Klein et al., 2008; Muntoni and Voit, 2004].
Syndromes, Genetics, and Molecular Basis
All of the cobblestone malformation syndromes with muscle involvement share the common feature of hypoglycosylated α-dystroglycan on skeletal muscle biopsy, and all of the known genes code for proteins known or suspected to be involved with glycosylation, particularly O-mannosylation of α-dystroglycan, which has led to the general term “dystroglycanopathy” for these and related CMD syndromes with normal brain structure [Brockington and Muntoni, 2005; Hewitt and Grewal, 2003; Muntoni et al., 2002; Ross, 2002]. Alpha- and β-dystroglycan are components of the dystrophin-associated glycoprotein complex that links the actin-associated cytoskeleton and extracellular matrix, and are encoded by the same precursor peptide. Alpha-dystroglycan is a highly glycosylated peripheral membrane protein that binds many extracellular matrix proteins through attached carbohydrate groups. In the dystroglycanopathies, especially the cobblestone-CMD group, glycosyl groups are absent or reduced, resulting in decreased binding of ligands such as laminin-2, agrin and perlecan in skeletal muscle, and neurexin in the brain [Barresi and Campbell, 2006].
Accordingly, all of the genes associated with cobblestone syndromes with muscle involvement code for proteins that regulate glycosylation, particularly O-mannosylation of α-dystroglycan. At least two of the non-CMD cobblestone syndromes are also congenital disorders of glycosylation [Cantagrel et al., 2010; Morava et al., 2009; Van Maldergem et al., 2008], and another is associated with a heavily glycosylated protein [Jin et al., 2007]. The cobblestone syndromes and associated genes are listed in Table 26-3, with estimated mutation frequencies shown in Table 26-4. Biochemical deficiency has been demonstrated in muscle for some of the proteins involved [Zhang et al., 2003].
Table 26-3 Cobblestone Syndromes, Inheritance and Genes
| Cobblestone Type and Syndrome | Inheritance | Genes |
|---|---|---|
| CLASSIC COBBLESTONE SYNDROMES WITH CMD | ||
| Walker–Warburg syndrome | AR | POMT2, POMGnT1, FKRP, FKTN, LARGE |
| Muscle-eye-brain disease | AR | POMT2, POMGnT1, FKRP, LARGE |
| Fukuyama CMD | AR | FKTN |
| Mental retardation, microcephaly, and CMD | AR | POMT1, POMT2 |
| Mental retardation, cerebellar cysts, and CMD | AR | FKRP |
| VARIANT COBBLESTONE SYNDROMES WITH NORMAL MUSCLE | ||
| Bilateral frontoparietal cobblestone malformation | AR | GPR56 |
| Dandy–Walker malformation with CDG* | AR | B4GALT1 |
| Debré-type cutis laxa | AR | ATP6V0A2 |
| CHIME-like syndrome | AR | SRD5A3 |
| CEDNIK syndrome | AR | SNAP29 |
AR, autosomal-recessive; CDG, congenital disorder of glycosylation; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis and keratoderma; CHIME, colobomas, heart defects, ichthyosiform dermatosis, mental retardation, and either ear defects or epilesy; CMD, congenital muscular dystrophy.
* Cobblestone cortical malformation has not been confirmed for this syndrome.
Walker–Warburg Syndrome
WWS consists of severe cobblestone malformation with a smooth surface resembling lissencephaly, as well as the most severe brainstem and cerebellar malformations of any of the cobblestone syndromes (see Figure 26-3C). Most patients have hydrocephalus, and approximately 25 percent have occipital cephaloceles [Dobyns et al., 1985, 1989]. All have profound mental retardation, epilepsy, and variable eye abnormalities, such as microphthalmia, congenital glaucoma with or without buphthalmos, Peter anomaly, iris hypoplasia, colobomas, cataracts, persistent hyperplastic primary vitreous, retinal dysplasia, and retinal nonattachment. All have CMD or congenital myopathy, with contractures and elevated serum levels of creatine kinase.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


