Malformations of the Central Nervous System
John H. Menkes
Harvey B. Sarnat
Laura Flores-Sarnat
Malformations of the brain and spinal cord may be genetically determined or acquired. The great majority of dysgeneses that occur early in gestation have a genetic basis, whereas those that begin late in gestation are more likely to be secondary to destructive lesions such as infarcts that interfere with development of particular structures. The distinction between atrophy, the shrinkage of a previously well-formed structure, and hypoplasia, the deficient development of a structure that never achieves normal size, is not always clear in degenerative processes or in those acquired lesions of fetal life in which an insult is imposed on a structure that is not yet fully formed. Examples are ischemic lesions in fetal brain associated with congenital cytomegalovirus infections and fetal degenerative diseases such as pontocerebellar hypoplasia and polymicrogyria, which develop in zones of relative ischemia that surround porencephalic cysts resulting from an occlusion of the middle cerebral artery incurred in fetal life. White matter infarcts in the cerebrum may destroy radial glial fibers and prevent normal migration of neuroblasts and glioblasts from the subventricular zone or germinal matrix (see Chapter 6).
Regardless of their cause, malformations are traditionally classified as disturbances in developmental processes. These are outlined in Table 5.8.
Whereas this type of classification retains validity for understanding the type of developmental process most disturbed, such as cellular proliferation or neuroblast migration, the new understanding of developmental genes and their role in the ontogenesis of the nervous system provides a new, complementary molecular genetic classification of early neurogenesis that recognizes the genetic regulation of development. An example of an attempt to use these new data to organize the thinking about developmental malformations of the brain is proposed in Table 5.9, a table that will undoubtedly undergo considerable revision in the coming years as more data become available.
TABLE 5.8 Traditional Classification of Central Nervous System Malformations as Disorders of … | |
|---|---|
|
Because the nervous system develops in a precise temporal as well as spatial sequence, it is often possible to assign a precise timing of a malformation, or at least to date the earliest time when the insult was first expressed. In most cases, an insult, whether caused by overexpression or underexpression of a developmental gene or caused by an ongoing acquired process such as a congenital viral infection or repeated episodes of ischemia, affects nervous system development over an extended period of time. Thereby, the insult involves processes that occur at various stages of development, not just at a single precise moment. As discussed in the Patterning of the Neural Tube:
Axes and Gradients of Growth and Differentiation section, developmental genes may serve as organizer genes early in ontogenesis and as regulator genes later on, thus involving various processes. Defective expression of SHH, for example, may result in holoprosencephaly because of its early effects on midline ventralization in the prosencephalon but may affect granule cell proliferation in the cerebellum as well; the timing of these two events is quite different.
Axes and Gradients of Growth and Differentiation section, developmental genes may serve as organizer genes early in ontogenesis and as regulator genes later on, thus involving various processes. Defective expression of SHH, for example, may result in holoprosencephaly because of its early effects on midline ventralization in the prosencephalon but may affect granule cell proliferation in the cerebellum as well; the timing of these two events is quite different.
TABLE 5.9 Proposed Molecular Genetic Classification of Malformations of Early Central Nervous System Development | |
|---|---|
|
The accounts that follow are traditional descriptions of major malformations of the human nervous system, but the new perspective of molecular genetic programming will be an integral part of the understanding of these disorders of development. Finally, it must always be recognized that just as no two adults, even monozygotic twins, are identical, no two fetuses are identical and no two cerebral malformations are identical. Individual biological variations occur in abnormal as well as in normal development, and allowance must be made for small differences while recognizing the principal patterns that denote pathogenesis.
The importance of disordered nervous system maturation in causing chronic abnormalities of brain function only recently has become fully apparent. Estimates suggest that 3% of neonates have major CNS or multisystem malformations (325), and 75% of fetal deaths and 40% of deaths within the first year of life are secondary to CNS malformations (326). Furthermore, 5% to 15% of pediatric neurology hospital admissions appear to be primarily related to cerebral and spinal cord anomalies (327). Genetic and nongenetic interactions are responsible for 20% of CNS malformations; monogenic malformations, whether autosomal or X-linked, account for 7.5% of malformations; chromosomal factors account for 6%; and well-delineated environmental factors, including maternal infections, maternal diabetes, irradiation, and drugs (e.g., thalidomide, valproic acid, methylmercury, excessive vitamin A or retinoic acid) account for at least another 3.5%. In the remainder, more than 60% of cases, the cause of the CNS malformation is uncertain (328,329,330). As more associations with specific genes and their defective expression become known, this number will undoubtedly become smaller.
EMBRYONIC INDUCTION DISORDERS (0 TO 4 WEEKS’ GESTATION)
Embryogenic induction disorders represent a failure in the mutual induction of mesoderm and neuroectoderm. The primary defect is a failure of the neural folds to fuse and form the neural tube (ectoderm), with secondary maldevelopment of skeletal structures enclosing the CNS (mesoderm). This process is called neurulation. In addition to the mesodermal notochord inducing the floor plate of the neural tube, the neural tube induces many non-neural structures of mesodermal origin. Craniofacial development is induced by the anterior neural tube and mediated by the migration of mesencephalic and prosencephalic neural crest tissue (293,294). This relation explains the midfacial hypoplasia in holoprosencephaly, the absence of calvarial bones and hypotelorism in anencephaly, and hypertelorism in agenesis of the corpus callosum and in many genetic syndromes such as Noonan syndrome (298,300).
Defects range from anencephaly to sacral meningomyelocele in the cephalic to caudal direction of the neural tube, and from holoprosencephaly to craniospinal rachischisis (midline posterior splitting of skull and vertebral column) in the anterior to posterior direction. For convenience, they are divided into dorsal (posterior) and ventral (anterior) midline defects. The former is named dysraphism to indicate the persistent continuity between posterior neuroectoderm and cutaneous ectoderm. Midline cerebral malformations with dysmorphic facies, such as holoprosencephaly, are not in this category. Some dorsalizing genes, expressed in the dorsal part of the neural tube (e.g., ZIC2, several PAX genes, the BMP family), may cause CNS malformations without dysraphism, however, when mutations occur. Ventral midline defects that involve more structures than just the neural tube are called faciotelencephalopathy to connote the noncleavage of the ventral neural tube, cephalic mesoderm, and adjacent foregut entoderm.
Dorsal Midline Central Nervous System–Axial Skeletal Defects: Dysraphism
A large number of midline anomalies occur, and the chance of several midline defects occurring conjointly is greater than the product of their individual occurrence.
Anencephaly
Anencephaly is the paradigm of the various dysraphic disorders. Although affected infants rarely survive early infancy, insight into the mechanics of neural ontogenesis provided by this disorder is enormous.
Pathogenesis and Pathology
Both genetic predisposition and environmental insults are responsible for the condition. The defect is time specific in that the insult probably occurs after the onset of neural fold development (16 days) but before closure of the anterior neuropore (24 to 26 days). The stimulus is nonspecific because a variety of insults have been implicated. These include drugs (330), infections (331), chemical disorders such as maternal diabetes or folic acid deficiency (332), and irradiation (333). Whatever the actual teratogenic stimulus might be, it induces four basic defects: (a) A
defective notochord and prechordal mesoderm (the notochord proper extends rostrally only to the midbrain) causes failure of the cephalic neural folds to fuse into a neural tube. (b) Failure of development of the meninges and cranial bones exposes the brain to amniotic fluid, with subsequent encephaloclastic degeneration of forebrain germinal cells. (c) Paraxial mesoderm fails to differentiate into well-formed somites and hence into sclerotomes, the latter being the primordium for the base of the skull and vertebrae. (d) A failure of prosencephalic and mesencephalic neural crest formation and migration results in midfacial hypoplasia with hypotelorism resembling that of holoprosencephaly and failure of formation of the meninges over the forebrain and membranous bone of the cranial vault (298,300). Thus, mutual induction between the germ layers fails at time-specific stages, resulting in deformities of both nervous tissue and supporting axial and membranous bone (300,334). Genetic factors are suspected in many cases, but no specific gene or its locus has yet been identified.
defective notochord and prechordal mesoderm (the notochord proper extends rostrally only to the midbrain) causes failure of the cephalic neural folds to fuse into a neural tube. (b) Failure of development of the meninges and cranial bones exposes the brain to amniotic fluid, with subsequent encephaloclastic degeneration of forebrain germinal cells. (c) Paraxial mesoderm fails to differentiate into well-formed somites and hence into sclerotomes, the latter being the primordium for the base of the skull and vertebrae. (d) A failure of prosencephalic and mesencephalic neural crest formation and migration results in midfacial hypoplasia with hypotelorism resembling that of holoprosencephaly and failure of formation of the meninges over the forebrain and membranous bone of the cranial vault (298,300). Thus, mutual induction between the germ layers fails at time-specific stages, resulting in deformities of both nervous tissue and supporting axial and membranous bone (300,334). Genetic factors are suspected in many cases, but no specific gene or its locus has yet been identified.
Studies of human embryos suggest that the splitting of an already closed neural tube might account for anencephaly and other dysraphic conditions (335). Gardner and Breuer argued not only that dysraphic states are a consequence of neural tube rupture after closure, but also that a number of associated non-neural anomalies, including asplenia, renal agenesis, and tracheoesophageal fistula, result from the damage of primordia of other organs by the overdistended neural tube (336). Osaka and coworkers discounted these theories on the basis that in human embryos dysraphism can be observed before completion of neural tube closure (337,338). Muscle differentiates normally in anencephaly despite disruption of motor innervation, suggesting that motor innervation occurs after muscle development and, therefore, after embryogenesis and neural tube closure (339). A primary defect of neural crest could explain many of the non-neurologic features of anencephaly (300).
Examination of the nervous system shows the spinal cord, brainstem, and cerebellum to be small. Descending tracts within the spinal cord, particularly the corticospinal tract, are absent. Above the midbrain, glial and vascular tissue with remnants of midbrain and diencephalon exist. Sometimes the basal telencephalic nuclei are partially or even fully formed (73). The pituitary is absent, with secondary adrenal hypoplasia. The optic nerves are absent but the eyes are normal, indicating that the anterior cephalic end of the neural tube, whence the optic vesicles spring, closed and diverticulated properly.
In addition to the primary defect of development, anencephaly involves an important encephaloclastic component (73,300). Because neural tissue is directly exposed to amniotic fluid, which is caustic, a progressive destruction of neural tissue and a compensatory proliferation of small blood vessels occurs to create the area cerebrovasculosa in the nubbin of tissue representing the residual prosencephalon. A poorly organized network of thin-walled vascular channels of variable size that are not mature capillaries, arterioles, or venules is enmeshed with glial processes and scattered (haphazardly oriented neurons and neuroblasts, lacking recognizable architecture as either nuclei or laminated cortex). Anencephaly is therefore difficult to analyze histopathologically because of the simultaneous presence of both primary dysplastic and secondary destructive processes.
The calvarium fails to develop, and the frontal and parietal bones are partially absent; the rostral half of the occipital bone is membranous and also is deficient, but the posterior part, not of neural crest origin, is preserved. Malformations of the foramen magnum and cervical vertebrae are frequent. The reduced forehead and relatively large ears and eyes lend a froglike appearance to the face; facial structures are developed, but midfacial hypoplasia with hypotelorism occurs in some cases (300), and there is an occasional lateral cleft lip or palate.
Some authors use the term aprosencephaly for cases in which the calvarium is intact, in distinction from atelencephaly, in which the cranium is open (340,341). In exencephaly, a rare condition, the membranous bones of the cranial vault are absent and the preserved but disorganized brain is covered by vascular epithelium. The condition is believed to be a stage in the development of anencephaly, with more complete destruction of the exposed brain being a matter of time (342).
Epidemiology
Anencephaly is the most common major CNS malformation in the West (343). The incidence of this malformation differs in various parts of the world. It is high in Ireland, Scotland, and Wales and low in Japan. The incidence of anencephaly and of neural tube defects in general is very high in northern but not southern China (344). It is high also in Mexico near the Texas border (345). Other areas of high incidence include Egypt, the Arabian subcontinent, and New Zealand. As a rule, the incidence increases with increasing maternal age and decreasing socioeconomic status.
The rate of anencephaly as well as that of the other neural tube defects has declined. In the 1960s, the incidence ranged from 0.65 per 1,000 births in Japan to more than 3 in 1,000 in the British Isles, with a maximum incidence of 8 in 1,000 occurring in Ireland in 1960. Prior peaks in incidence had been recorded during the years of 1929 through 1932 and 1938 through 1941. Since then, there has been a steady decline in both the United States and the United Kingdom. Between 1971 and 1989, the annual rate of various forms of spina bifida fell from 2 in 1,000 to 0.6 in 1,000 (346), with a relative increase in the proportion of spina bifida to anencephaly (347). In part, this decline reflects the widespread use of antenatal screening, but other factors,
notably the correction of maternal vitamin deficiency, also might be responsible (346,348).
notably the correction of maternal vitamin deficiency, also might be responsible (346,348).
Anencephaly is seen 37 times more frequently in female than in male newborns (349). The recurrence rate in families with an affected child is 35%, although almost 10% of siblings of anencephalics have major anomalies of neural tube closure: anencephaly, spina bifida cystica, and encephalocele (350). The transmission appears to be matrilineal. No relationship to consanguinity is evident, nor to concordance in monozygotic twins, and the recurrence rate for a maternal half-sibling is the same as for a full sibling. These factors weigh against a simple polygenetic inheritance pattern and are more consistent with the interaction of genetic and environmental factors (351).
Clinical Manifestations
Anencephalic patients do not survive infancy. During their few weeks of life, they exhibit slow, stereotyped movements and frequent decerebrate posturing. Head, facial, and limb movements can be spontaneous or pain induced. The Moro reflex and some brainstem functions and automatisms, such as sucking, rooting, and righting responses, are present and are more readily and more reproducibly elicited than in healthy infants. The bowing reflex, which occasionally can be demonstrated in healthy premature infants of 7 months’ gestation, is invariably present in anencephalics (352) (see Introduction chapter). Seizures have been observed in anencephalic infants, an indication that some types of neonatal seizures originate in the deeper structures of the brain (353).
The presence of anencephaly and other open neural tube defects can be predicted by measuring α-fetoprotein (AFP) in amniotic fluid or maternal serum. AFP is the major serum protein in early embryonic life, representing 90% of total serum globulin. It is a fetus-specific α1-globulin that is probably involved in preventing fetal immune rejection; it is produced first by the yolk sac and later by the fetal liver and gastrointestinal tract. It normally passes from fetal serum into fetal urine and then into amniotic fluid. Because of a substantial leak of fetal blood components directly into amniotic fluid, AFP concentrations in amniotic fluid and maternal serum AFP levels are elevated in anencephaly and in open spina bifida or cranium bifidum (354).
Normal AFP in adult serum is less than 10 ng/mL. In normal maternal serum and amniotic fluid, it ranges from 15 to 500 ng/mL. At 15 to 20 weeks’ gestation, an AFP concentration of 1,000 ng/mL or greater strongly suggests an open neural tube defect, and the current screening of serum detects 79% of cases of open spina bifida at 16 to 18 weeks (355). Determining gestational age is critical, however, because normal AFP concentration varies considerably with fetal age, peaking between 12 and 15 weeks’ gestation. Amniotic fluid AFP screening is more reliable, detecting 98% of open spina bifida cases (356). The amniotic fluid must be assessed for contamination by fetal hemoglobin, which complicates amniocentesis, because a 200:1 AFP gradient exists between fetal serum and amniotic fluid. The reliability of ultrasonography depends on the experience of the operators; in good hands, the procedure is more than 99% specific (355). False-positive results are obtained in a variety of unrelated conditions, principally in the presence of multiple pregnancies, threatened abortion or fetal death, or when an error is made in dating the pregnancy. Amniotic fluid AFP obtained between 15 and 20 weeks’ gestation is most specific (356); however, closed neural tube defects such as skin-covered lipomyelomeningoceles, encephaloceles, and meningoceles go undetected. These lesions constitute between 5% and 10% of total neural tube defects (357,358).
Mothers who have borne one or more children with neural tube defects, spinal dysraphism, or multiple vertebral anomalies; who have a family history of any of these disorders; or who are surviving patients with spina bifida are at risk for bearing children with neural tube defects and should undergo screening.
Supplementation of the maternal diet with folic acid or with a multivitamin preparation that contains folic acid even before conception has been proposed to prevent neural tube defects. An extensive, controlled British study indicated that the recurrence rate of neural tube defects can be reduced sharply by folic acid supplementation (359). A multivitamin cocktail including folic acid, ascorbic acid, and riboflavin, given from at least 28 days before conception up to the second missed menstrual period, reduced the recurrence rates for neural tube defects from 4.2% to 0.5% in mothers with a previous neural tube defect pregnancy and from 9.6% to 2.3% in mothers who had given birth to two or more offspring with neural tube defects (360,361). These findings were duplicated in an American study, which showed that a vitamin supplement including 0.8 mg of folic acid, started 1 month before conception reduced significantly the incidence of neural tube defects (362,363). The reason for the apparent effect of folic acid is unclear, and the significance of these findings must be evaluated in the light of the declining incidence of neural tube defects in areas where no vitamin supplementation is used (346,364,365). Mice with a mutation of the Cart1 gene develop acrania and meroanencephaly, and this can be prevented by prenatal folic acid treatment (94).
Meningomyelocele (Spina Bifida) and Encephalocele (Cranium Bifidum)
As the older names imply, spina bifida and cranium bifidum share a failure of bone fusion in the posterior midline of the skull (cranium bifidum) or the vertebral column (spina bifida). The result is a bony cleft through which the meninges and varying quantities of brain or spinal cord tissue protrude. In cranium bifidum, the neural herniation is
termed encephalocele and can consist of brain parenchyma and meninges or only of meninges. These form the wall of a saclike cyst filled with cerebrospinal fluid (CSF). Posterior encephaloceles may contain only supratentorial structures, only posterior fossa structures, or both. In spina bifida, the herniation is called meningocele or meningomyelocele, depending on whether the meninges herniate alone or together with spinal cord parenchyma and nerve roots. The traditional names spina bifida and cranium bifidum are now less frequently used than in the older literature because of the recognition that the bony cleft may not be the primary defect in all cases, but instead that the pathogenesis may involve neural induction of mesodermal tissues in the dorsal midline, including leptomeninges, dura mater, and bone.
termed encephalocele and can consist of brain parenchyma and meninges or only of meninges. These form the wall of a saclike cyst filled with cerebrospinal fluid (CSF). Posterior encephaloceles may contain only supratentorial structures, only posterior fossa structures, or both. In spina bifida, the herniation is called meningocele or meningomyelocele, depending on whether the meninges herniate alone or together with spinal cord parenchyma and nerve roots. The traditional names spina bifida and cranium bifidum are now less frequently used than in the older literature because of the recognition that the bony cleft may not be the primary defect in all cases, but instead that the pathogenesis may involve neural induction of mesodermal tissues in the dorsal midline, including leptomeninges, dura mater, and bone.
Spina bifida occulta is a minor fusion failure of the posterior vertebral arches unaccompanied by herniation of meninges or neural tissue. Spina bifida cystica collectively designates meningocele, meningomyelocele, and other cystic lesions (Fig. 5.1). Similarly, in the head, cranium bifidum comprises meningocele, a herniation of meninges containing only CSF, and the more commonly occurring encephalocele, in which the sac contains neural and glial tissue. Rachischisis refers to a severe condition with an extensive defect of the craniovertebral bone with exposure of the brain, spinal cord, and meninges. Myeloschisis is another defect in the tissues over the lower spinal cord. Neural tissue is exposed at the surface as a flat, red lesion with a velvety appearance over the sacral region, without protruding as a myelomeningocele sac.
Pathogenesis
Spina bifida and cranium bifidum are not only disorders of induction; they also are associated with major abnormalities of cellular migration and secondary mechanical deformities of the nervous system. The continuity between neural and cutaneous ectodermal derivates is regarded as evidence that the primary defect is in the neural tube closure (366,367). Based on studies with embryos of mutant mice with genetically abnormal neurulation and a sacral neural tube defect, McLone and Naiditch (368) proposed a unified theory for the development of the associated anomalies that incorporates some of the prior observations of Padget (334).
According to these authors, the initial event is a failure of the neural folds to close completely, leaving a dorsal myeloschisis. This is followed by a failure of the normal, transient occlusion of the central cavity of the spinal cord. These two events result in the escape of CSF into the amniotic cavity and a collapse of the primitive ventricular system. The failure of the primitive cranial ventricular system to distend results in a posterior fossa that is too small to accommodate the growing cerebellum and leads to upward and downward herniation of the structures within the posterior fossa. Additionally, the failure of the normal distention of the ventricular system leads to inadequate support for the normal outward migration of neuroblasts and a failure to maintain the normal pattern of ossification in the calvarium.
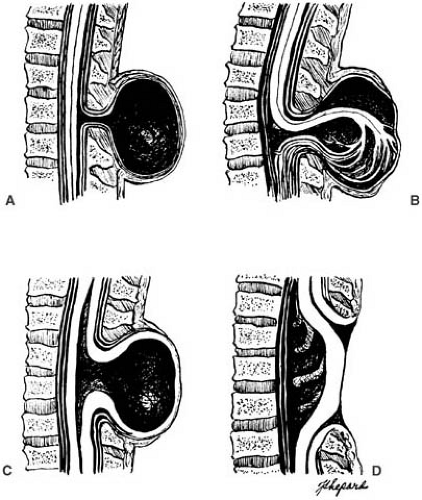 FIGURE 5.1. Drawings of various forms of spinal dysraphic lesions (spina bifida cystica). A: Meningocele. Through the bony defect (spina bifida), the meninges herniate and form a cystic sac filled with spinal fluid. The spinal cord does not participate in the herniation and might or might not be abnormal. B: Myelomeningocele. The spinal cord is herniated into the sac and ends there or can continue in an abnormal way further downward. C: Myelocystocele or syringomyelocele. The spinal cord shows hydromyelia; the posterior wall of the spinal cord is attached to the ectoderm and is undifferentiated. D: Myelocele. The spinal cord is araphic; a cystic cavity is in front of the anterior wall of the spinal cord. (From Benda CE. Developmental disorders of mentation and cerebral palsies. New York: Grune and Stratton, 1952. With permission.) |
Marín-Padilla proposed that the primary defect is a limited injury to the primitive streak and primitive node, which impairs local growth of skeletal elements, which in turn interferes with closure of the neural tube (369). Although the specific genes involved have not been identified, this hypothesis may be expanded to invoke a mechanism of failed expression of one or more organizer genes during the primitive streak and neural placode stages of early ontogenesis.
These defects are time specific, which is why the most common sites for the lesion in surviving children are either lumbosacral or occipital, these being the last levels at which neural tube closure normally occurs. The initiation
of a defect at an earlier stage leads to a more extensive defect, which is incompatible with survival. In the same way, a simple meningocele results when the insult occurs after the spinal cord has formed, whereas a myelomeningocele arises from an earlier insult, which must occur before closure of the posterior neuropore (i.e., before 26 to 28 days’ gestation) (Table 5.10) (370).
of a defect at an earlier stage leads to a more extensive defect, which is incompatible with survival. In the same way, a simple meningocele results when the insult occurs after the spinal cord has formed, whereas a myelomeningocele arises from an earlier insult, which must occur before closure of the posterior neuropore (i.e., before 26 to 28 days’ gestation) (Table 5.10) (370).
TABLE 5.10 Timetable of Human Central Nervous System Ontogenesis | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The cause of these anomalies is unknown. As is the case for anencephaly, it is likely that genetic defects, probably at more than one locus, interact with environmental factors to produce the varying dysraphic conditions. Spinal dysraphic lesions are among the most common anomalies of the nervous system. As with anencephaly, the incidence is highest in Ireland and lowest in Japan and also is influenced by season, socioeconomic status, gender, ethnicity, and such maternal factors as parity, age, prior offspring with neural tube defects, and maternal heat exposure (371). Recurrence rates for mothers who have previously given birth to a child with an open neural tube defect are 1.5% to 2.0%, and for mothers with two affected children, the recurrence rate is 6% (372). The recurrence risk is also higher than normal if close relatives are affected. Less is known about the epidemiology of cranium bifidum, the incidence of which is approximately 1/10 that of spina bifida cystica (341,373).
Pathology
Meningomyelocele (Spina Bifida Cystica).
Of the defects collectively termed spina bifida cystica, 95% are myelomeningoceles and 5% are meningoceles. Locations of the defect in liveborn infants are depicted in Table 5.11.
A lumbar or lumbosacral defect is most common; it corresponds to the site of the posterior neuropore closure. Cervical lesions are the least frequent posterior defects. Anterior midline defects of the vertebral arches are uncommon and constituted less than 0.5% of cases in the experience of Matson (373). Approximately 100 anterior sacral meningoceles have been reported, the majority in female patients (374). These conditions should be differentiated from spinal meningeal malformations, which can occur in isolation or in association with systemic malformations (375). Spinal meningeal malformations are relatively common in patients with the various mucopolysaccharidoses and in neurofibromatosis.
A lumbar or lumbosacral defect is most common; it corresponds to the site of the posterior neuropore closure. Cervical lesions are the least frequent posterior defects. Anterior midline defects of the vertebral arches are uncommon and constituted less than 0.5% of cases in the experience of Matson (373). Approximately 100 anterior sacral meningoceles have been reported, the majority in female patients (374). These conditions should be differentiated from spinal meningeal malformations, which can occur in isolation or in association with systemic malformations (375). Spinal meningeal malformations are relatively common in patients with the various mucopolysaccharidoses and in neurofibromatosis.
TABLE 5.11 Site of Lesion of Spina Bifida Cystica | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
Cervical and thoracic meningoceles have narrow bases and are usually not associated with hydrocephalus. By contrast, 90% or more of lumbosacral myelomeningoceles are accompanied by Chiari type II malformations and hydrocephalus. As originally described by Chiari, type I malformations consist of heterotopic, downwardly displaced cerebellar tissue in the absence of space-occupying lesions other than hydrocephalus. Type III malformations consist of cervical spina bifida accompanied by a cerebellar encephalocele. Type IV is a heterogeneous variant in which the cerebellum and brainstem remain in their entirety within the posterior fossa but the cerebellum is small (376,377). Type IV is now an obsolete term of historical interest and is redesignated cerebellar hypoplasia.
In 88% of children with lumbar or lumbosacral meningomyeloceles, the spinal cord demonstrates abnormalities in the cervical region (Table 5.12) (366). The majority of instances involve hydrosyringomyelia; less often, diplomyelia or winged and dorsally slit cords are present (378,379). In greater than 70% of cases, the medulla overrides the cervical cord dorsally, in association with type II Chiari malformation (Figs. 5.2 and 5.3). Chiari II malformation is the most constant accompanying feature of lumbosacral meningomyeloceles and is present in nearly all cases. Of patients with spina bifida cystica, 70% show defects in the posterior arch of the atlas, which is bridged by a firm fibrous band, suggesting that congenital atlantoaxial dislocation is a mild expression of an induction disorder (380). Examination of the parenchyma of the spinal cord reveals atrophic or poorly developed ventral horn cells, absent or abnormal corticospinal and ascending sensory tracts, incomplete posterior horns, and exceedingly small and deranged ventral and dorsal root fibers. These changes result in muscle denervation during fetal life and ultimately produce limb deformities and joint contractures.
Defects of cellular migration in the cerebral hemispheres are extremely common (see Table 5.12). These include gray matter heterotopia, schizencephaly, gyral anomalies, agenesis of the corpus callosum, and mesodermal ectopia (366,381,382).
A number of mesodermal lesions accompany the ectodermal defects. In addition to the spinal canal being widened and the posterior arches being malformed, the vertebral bodies can be misshapen with resulting kyphosis or scoliosis. Rib anomalies are common. Mesodermal dysplasia of the skull produces defects in the membranous bones of the calvarium, a condition termed lacunar skull or craniolacunia (Lückenschadel). This peculiar, honeycombed appearance of the skull is seen in some 85% of patients with the Chiari type II malformation. The skull changes are transient and disappear in the first few months after birth. They are probably the result of a defect in membranous bone formation and are not secondary to in utero intracranial hypertension, as is often stated (383). The lattice pattern in the inner table of the cranium does not correspond to cerebral convolutions. Lacunar skull also can be seen, rarely, in neonates with normal brains, no midline defects over the spine or head, and no neurologic symptoms (384).
TABLE 5.12 Central Nervous System Anomalies Associated with Meningomyelocele, Hydrocephalus, and Arnold-Chiari Malformation | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
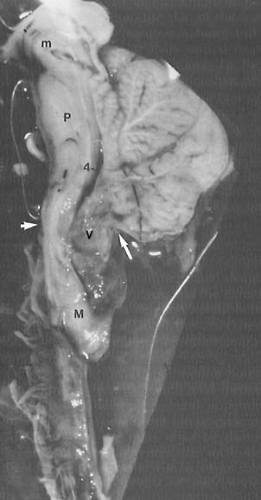 FIGURE 5.2. Chiari type II malformation. Sagittal section through the cerebellum and brainstem in a newborn boy. Anterior is to the reader’s left. Arrows mark the location of the foramen magnum. The medulla (M) protrudes below the foramen magnum into the cervical spinal cord canal to overlap the cervical spinal cord. The medulla buckles dorsally to form a kink. The cerebellar vermis (V) is indented by the posterior lip of the foramen magnum. The fourth ventricle (4) is elongated, and the midbrain (m) is beaked. The pons (P) is demonstrated also. (From Naidich TP, McLone DG, Fulling KH. The Chiari II malformation: part IV. The hind-brain deformity. Neuroradiology 1983;25:179–197. With permission.) |
Deformities of the lower extremities are common and are of two types. In the first type, the various clubfoot and rocker-bottom foot deformities result from the unopposed action of the intrinsic foot muscles or the muscles at the ankle joint. In the second type, the deformities are positional; they result from intrauterine pressure on the paralytic limbs.
Other anomalies accompany myelodysplasia with a greater-than-normal incidence. These include intestinal malformations (e.g., duodenal atresia, pyloric stenosis, anal stenosis), renal anomalies, notably renal agenesis, urogenital defects, cardiac malformations, and tracheo-esophageal fistulas.
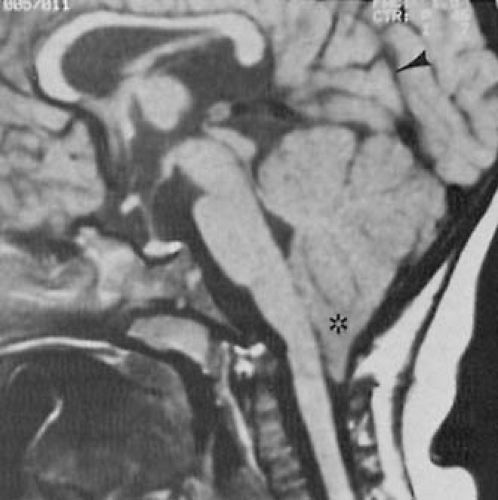 FIGURE 5.3. Chiari type II malformation. Magnetic resonance imaging study. The fourth ventricle and aqueduct are stretched only slightly. Cerebellar heterotopia includes both the inferior vermis and tonsil (asterisk). The tentorial opening is wide, and vertical orientation is seen at line of attachment along the straight sinus (black arrowhead). (Courtesy of Dr. Taher El Gammal, Department of Radiology, Medical College of Georgia, Augusta, GA, and the American Society of Neuroradiology.) |
Spina Bifida Occulta.
In spina bifida occulta, no herniation of the meninges is present and the skin of the back is completely epithelialized, although always showing some abnormality such as a nevus, dermal sinus, and dimple (35%), an underlying lipoma (29%), or a hirsute area (372). Radiography reveals a variety of deformities, the most common of which are widening of the spinal canal, fusion of the vertebral bodies, fused and malformed laminae, spina bifida, and, sometimes, a midline bone mass within the spinal canal. These skin and bone abnormalities are indications that the cord and nerve roots are malformed also. There may be a localized doubling of the cord (diplomyelia), a sagittal splitting of the cord (diastematomyelia), absent or adherent nerve roots, or an intradural lipoma attached to the cord. Abnormalities of the filum terminale, notably a shortening, which gives the appearance of a lengthening of the cord but actually results from a failure of the cord to dedifferentiate during early embryonic life, were seen in 24% of patients in Anderson’s series (383). Duplication of the spinal cord or portions of it, such as the central canal, is associated, as discussed in the Families of Developmental Genes of the Central Nervous System section, with upregulation of an early ventralizing influence of a gene such as Sonic hedgehog.
These lesions must be recognized because they can cause progressive loss of neural functioning during the childhood growth spurt. In many cases, operative intervention to free the cord or nerves is indicated to prevent
further damage or prophylactically to avoid such damage. One distinction of occult dysraphism is that it never seems to be accompanied by a Chiari II malformation. However, its genetic origins in familial cases are the same as those of spina bifida cystica, so that both types of spina bifida can occur in the same family. The chances that parents of a child with spina bifida occulta could have another offspring with spina bifida cystica are the same as when the proband has spina bifida cystica (385).
further damage or prophylactically to avoid such damage. One distinction of occult dysraphism is that it never seems to be accompanied by a Chiari II malformation. However, its genetic origins in familial cases are the same as those of spina bifida cystica, so that both types of spina bifida can occur in the same family. The chances that parents of a child with spina bifida occulta could have another offspring with spina bifida cystica are the same as when the proband has spina bifida cystica (385).
Cranium Bifidum.
Several types of simple midline or paired paramedian skull defects are grouped under the term cranium bifidum occultum. These include the persistence of wide fontanelles and parietal foramina (386). Persistently large foramina are seen in families, and the condition is sometimes transmitted as an autosomal dominant trait with the gene probably being located on the short arm of chromosome 11 (387). The condition has been termed Caitlin marks, named after the family in which it was described. Excessively large anterior and posterior fontanelles as hypomineralization of the cranium also occurs in hypophosphatasia; serum calcium and phosphate should be measures in such patients. Parietal foramina are generally asymptomatic, although they have been reported to be accompanied by a seizure disorder. The radiographic changes in the various congenital anomalies of the skull are reviewed by Kaplan and colleagues (388). Persistence of the fontanelle is sometimes accompanied by cleidocranial dysostosis, Marden-Walker syndrome, Schinzel-Giedion syndrome, and several other malformation syndromes (389).
Cranium bifidum (cephalocele, encephalocele) is a much more serious condition. Like anencephaly, it has been postulated to represent a defect in the closure of the anterior neuropore. Hoving and coworkers, however, proposed that the underlying defect is a disturbance in the separation of neural and surface ectoderm (390). Marín-Padilla suggested that it results from a deficiency in local growth of the basicranium, with the timing of the insult and the amount of damage to mesodermal cells determining whether the result is anencephaly, Chiari II malformation, or cranium bifidum (369).
The incidence of cranium bifidum is approximately 1/10 that of spina bifida cystica. In the Western world, approximately 85% of these lesions are dorsal defects involving the occipital bone. Parietal, frontal, or nasal encephaloceles are far less common. In Asia, the majority of encephaloceles are anterior and involve the frontal, nasal, and orbital bones (373,391,392). The lesions of cranium bifidum, regardless of whether they are a meningocele or contain neural tissue, and consequently are an encephalomeningocele, are usually classified together as encephaloceles. As with myelomeningoceles, the sac can be covered by partially transparent abnormal meninges, but in most lesions, the herniation is fully epithelialized with either dysplastic or normal skin. Cutaneous abnormalities are frequent and consist of port wine stains, abnormal patterning of scalp hair, a hairy nevus over the posterior lumbosacral region, and, occasionally, excessive amounts of subcutaneous lipomatous tissue.
In the series of Simpson and coworkers, 34% of occipital meningoencephaloceles contained only cerebral tissue, 21% had cerebral and cerebellar tissue, and 37% had nodules of glial cells and dysplastic neural tissue (393). In 5% the sac contained cerebellar tissue only. Some of these infants would represent the Chiari III malformation. MRI is invaluable in determining the contents of the encephalocele (341).
Lorber and Schofield reported 147 cases of posteriorly located encephaloceles (392). Of this group, one-fifth were cranial meningoceles and the remainder were encephalomeningoceles. Of those patients who survived into childhood, 25% of those who harbored a meningocele and 75% of those with an encephalomeningocele exhibited mental retardation. All patients with microcephaly had neural tissue within the sac, and all exhibited mental retardation. The presence of neural tissue in the sac usually was associated with malformations of the hindbrain or, less often, with holoprosencephaly or agenesis of the corpus callosum (394). In the series of Lorber and Schofield, 16% of patients with encephaloceles had other anomalies, including myelomeningocele, cleft palate, congenital malformations of the heart, and Klippel-Feil syndrome (392). Hydrocephalus was present in more than 59% of patients and was more common in those with encephalomeningoceles. In a small proportion of patients with encephaloceles, the condition is part of a known syndrome. These were listed by Cohen and Lemire (395). Meckel-Grüber syndrome is probably the most common of these. It is an autosomal recessive condition with its gene mapped to chromosome 17q21–q24. It is characterized by an occipital encephalocele, holoprosencephaly, the Dandy-Walker syndrome, orofacial clefts, microphthalmia, polydactyly, polycystic kidneys, and cardiac anomalies (396).
In Western countries, only a small fraction of encephaloceles are located anteriorly. Most of these patients are otherwise completely healthy neurologically, and hydrocephalus is rare. The only associated CNS malformations are agenesis or lipomas of the corpus callosum (397). Midline frontal encephaloceles may be due to defective migration of the vertical sheet of prosencephalic neural crest (300). Anterior encephaloceles located at the cranial base often cause no external physical abnormalities, or they might be accompanied by such midline defects as hypertelorism, cleft lip, and cleft palate. An encephalocele presenting a mass that obstructs the nares can be mistaken for a nasal polyp. Its removal can result in a persistent CSF leak and meningitis.
On examination, the encephalocele is usually fully epithelialized, although the skin can be dysplastic. Its size
ranges from the insignificant to a sac that can rival the calvarium in size. Pedunculate lesions are less likely to contain neural tissue than sessile lesions. Transillumination can provide an indication of neural tissue in the sac; however, neuroimaging studies are definitive and detect associated CNS abnormalities.
ranges from the insignificant to a sac that can rival the calvarium in size. Pedunculate lesions are less likely to contain neural tissue than sessile lesions. Transillumination can provide an indication of neural tissue in the sac; however, neuroimaging studies are definitive and detect associated CNS abnormalities.
Meningocele.
A meningocele, by definition, represents the herniation of only the meninges through the defective posterior arches; the sac does not contain neural elements. Meningoceles account for less than 5% of patients with spina bifida cystica (398). Meningomyelocele must be differentiated from meningocele because the prognoses are vastly different. The meningomyelocele sac contains, in addition to cutaneous and subcutaneous tissues, meninges, fragments of bone, cartilage, and fibrous tissue, and neural elements. The neural tissue includes nerve roots and sometimes dysplastic spinal cord fragments and poorly differentiated neuroepithelium. An infant with a meningocele has little or no associated CNS malformation, rarely develops hydrocephalus, and usually has a normal neurologic examination. The anatomic distribution of meningoceles is the same as for myelomeningoceles. In general, meningoceles are fully epithelialized and tend to be more pedunculated than sessile lesions. Occasionally, a myelomeningocele is differentiated from a meningocele only at the time of operative repair. Some meningoceles contain a significant component of adipose tissue and are designated lipomeningoceles. These have a poorer long-term prognosis because the lipomatous portion often envelops nerve roots of the cauda equina and is not easily dissected from the roots at the time of surgery without sacrificing roots and creating a major neurologic deficit in the lower limbs and some visceral organs such as the urinary bladder.
Clinical Manifestations
Meningomyelocele (Spina Bifida Cystica).
At birth, spina bifida cystica can assume a variety of appearances. These range from complete exposure of neural tissue to a partially epithelialized membrane. Most often, a saclike structure is located at any point along the spinal column. Usually, the sac is covered by a thin membrane that is prone to tears, through which the CSF leaks. Of defects, 95% are myelomeningoceles and produce neurologic dysfunction corresponding to their anatomic level (399).
The lumbosacral region is the site of 80% of meningomyeloceles (see Table 5.11). These produce a variety of conus, epiconus, and cauda equina syndromes (Table 5.13). When the lesion is below L2, the cauda equina bears the brunt of the damage. Children exhibit varying degrees of flaccid, areflexic paraparesis and sensory deficits distal from the dermatome of L3 or L4. The sphincter and detrusor functions of the bladder are compromised and dribbling incontinence occurs. An absent or unilateral anal skin reflex and poor tone of the rectal sphincter are often apparent and can result in rectal prolapse. If the lesion is located at the thoracolumbar level or higher, the anal tone is often normal and the bladder is hypertonic. Lesions below S3 cause no motor impairment but can result in bladder and anal sphincter paralysis and saddle anesthesia involving the dermatomes of S3 through S5. Electromyographic studies and nerve conduction velocities obtained in the lower extremities of affected newborns suggest that the paralysis is the outcome of a combined upper and lower motor neuron lesion (400). Upper motor neuron lesions that result from involvement of the corticospinal tracts, however, usually are obscured by the more severe involvement of the nerve roots, cauda equina, and ventral horn cells.
TABLE 5.13 Neurologic Syndromes with Myelomeningoceles | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Cauda equina lesions produce muscular denervation in utero, resulting in joint deformities of the lower limbs. These are most commonly flexion or extension contractures, valgus or varus contractures, hip dislocations, and lumbosacral scoliosis. The expression of the contracture depends on the extent and severity of dermatome involvement.
Hydrocephalus associated with type II Chiari malformation complicates more than 90% of lumbosacral myelomeningoceles (401). It is manifest at birth in 50% to 75% of cases. In approximately 25% of infants with this condition, the head circumference is below the fifth percentile (373). In these infants and in the group whose head circumference is normal at birth, the ventricles are dilated at birth. This finding suggests that hydrocephalus almost always precedes operative closure of the myelomeningocele sac (402). Hammock and coworkers proposed that some of the infants with large ventricles but normal head circumference have normal-pressure hydrocephalus, and that these patients, like adults with this syndrome, might benefit from shunting procedures (403).
Clinical signs of progressive hydrocephalus accompanying a myelomeningocele include an abnormal
increase in head circumference, full fontanelle, spreading of sutures, hyper-resonant calvarial percussion note, dilated scalp veins, deviation of the eyes below the horizontal (setting sun sign), strabismus, and irritability. MRI studies have become the definitive diagnostic procedure for the evaluation of spina bifida cystica and the various other dysraphic conditions (381). They reveal the downward displacement of the stretched brainstem, with a kink between the medulla and the cervical spinal cord, and herniation of the cerebellar vermis. These findings are best seen on sagittal views. As a consequence of these malformations, CSF circulation is blocked at the level of the foramen magnum. Additionally, a significant incidence of hydromyelia of the cervical or thoracic spinal cord occurs (404). MRI and cine-MRI also can be used to display the patency of the aqueduct (405). Using MRI, El Gammal and coworkers found aqueductal stenosis in 40% of patients with myelomeningocele (404).
increase in head circumference, full fontanelle, spreading of sutures, hyper-resonant calvarial percussion note, dilated scalp veins, deviation of the eyes below the horizontal (setting sun sign), strabismus, and irritability. MRI studies have become the definitive diagnostic procedure for the evaluation of spina bifida cystica and the various other dysraphic conditions (381). They reveal the downward displacement of the stretched brainstem, with a kink between the medulla and the cervical spinal cord, and herniation of the cerebellar vermis. These findings are best seen on sagittal views. As a consequence of these malformations, CSF circulation is blocked at the level of the foramen magnum. Additionally, a significant incidence of hydromyelia of the cervical or thoracic spinal cord occurs (404). MRI and cine-MRI also can be used to display the patency of the aqueduct (405). Using MRI, El Gammal and coworkers found aqueductal stenosis in 40% of patients with myelomeningocele (404).
Other cerebral anomalies also have been described (366). These include microgyria and other types of cortical dysgenesis. These are frequently visualized by MRI studies but may be difficult to see if a deficit in cortical lamination is at the microscopic level, below the limit of resolution of imaging.
In the first few weeks or months of life, a small percentage of infants develop lower cranial nerve palsies and impaired brainstem function. This dysfunction is characterized by vocal cord paralysis with inspiratory stridor, retrocollis, apneic episodes, difficulty with feeding, and inability to handle secretions (406,407). Additionally, progressive spasticity of the upper extremities can develop (406). The reason for this progressive neurologic deficit is unclear. It might relate to compression of the cranial nerves and brainstem in the shallow posterior fossa. Downward pressure from inadequately controlled hydrocephalus has been suggested as an explanation for infants whose symptoms resolve with surgery. In others, brainstem hemorrhage, brainstem ischemia, or an underlying neuronal agenesis is present (406,408). Anterior sacral meningoceles are characterized by unremitting and unexplained constipation and a smooth pelvic mass. Their presence can be diagnosed by MRI studies.
Radiography of the spine reveals the extent of the nonfused vertebrae. The relationship between the cord segment and the vertebral bodies is abnormal. Although at birth the terminal segments of the normal cord lie between the vertebral bodies of T11 and L1, in infants with myelomeningocele the cord can extend as far down as L5 or even lower. The position of the spinal cord segments remains normal in the lower cervical and upper thoracic levels (409).
A dysraphic lesion that straddles the categories of occult and cystic spina bifida is the subcutaneous lipoma extending intradurally through a posterior vertebral arch defect to end within the substance of a low-lying conus medullaris. A more extreme example of this type of lesion is the lipomeningomyelocele (Fig. 5.4). This lesion can be included in either the cystic or the occult dysraphic category because a huge mass is evident with some lesions, whereas others are only a minimal deformity of the back. The mass is invariably located in the lumbosacral region. It can be midline or eccentric, is fully epithelialized, and frequently is associated with a cutaneous angioma, a hair patch, one or more dimples, or a sinus tract. In addition to the fatty tissue, the mass can be cystic or occasionally it can contain cartilage. These lesions are usually not associated with the Chiari malformations, hydrocephalus, or other CNS anomalies.
Though some infants with lumbosacral lipoma have no neurologic deficit, it is more common to find the lower lumbar or sacral segments affected, with resultant motor or sensory loss in the feet and bladder and bowel dysfunction. The quantity of subcutaneous lipomatous material varies. It extends through the defective posterior arches to become intimate with the low-lying and tethered conus medullaris. The dura is dysplastic and blends into the fatty tissue or forms cystic cavities filled with CSF.
Surgical intervention is advised at approximately age 3 months, not simply for cosmetic reasons, but, more important, to decompress and untether as far as possible the spinal cord, thus preventing progressive neurologic dysfunction. These patients require full orthopedic and urologic evaluation. A considerable proportion present with urologic symptoms, notably incontinence, soiling, and recurrent urinary tract infections (410).
Occasionally, a sacrococcygeal teratoma is mistaken for spina bifida cystica. Sacrococcygeal teratomas are only approximately 1/40 as frequent as spina bifida cystica, with a marked female preponderance. As the name implies, a sacrococcygeal teratoma is located in the sacrococcygeal region, whereas the lesion of spina bifida cystica is above the coccyx. Other than the breakdown of skin owing to tumor necrosis, no cutaneous abnormalities are seen. Deformities of the lower extremities and neurologic deficits are unusual, and radiography of the vertebral column is normal. Calcium deposits within the teratoma are seen in approximately one-third of patients. Imaging studies of the region confirm the diagnosis. A sacrococcygeal teratoma must be surgically removed in the first few days of life because the incidence of malignancy increases from 10% at birth to more than 50% by 2 months of age (411).
Malformations and infections of the genitourinary tract occur in up to 90% of newborns with spina bifida cystica, and renal disease is the most common cause of morbidity and mortality after age 3 years (412). Most commonly, a disturbance in bladder function is evident. One group, representing 33% of patients, shows more or less constant dribbling, and the bladder content is easily expressed manually. Direct cystometry reveals absent detrusor activity (413). In another, larger group of patients,
detrusor contractions are weak, but bladder emptying is inefficient, and outlet obstruction at the level of the external sphincter occurs. This obstruction is believed to result from impaired coordination between the detrusor and sphincter functions and reflects a lesion of the spinal cord between the pontine-mesencephalic center regulating the vesicourethral unit and the sacral area. Bladder sensation is intact in some children. This latter type of upper motor neuron defect results in a high incidence of bladder trabeculation, an elevated resting bladder pressure, and dilatation of the upper urinary tract, often reaching enormous proportions (414). Continence appears to depend more on preservation of detrusor activity than of sphincter function. Serial neurologic evaluations indicate that these abnormalities are not static, but tend to change, particularly during the first year of life. Some children with complete or nearly complete denervation of the external sphincter improve, whereas others deteriorate (415). When deterioration occurs in childhood, it most likely occurs in patients with dyssynergia with a small, trabeculated, noncompliant bladder (416). Persistent bacteriuria is seen in 50% of 2-year-old children, with hydronephrosis being found in 25% (417).
detrusor contractions are weak, but bladder emptying is inefficient, and outlet obstruction at the level of the external sphincter occurs. This obstruction is believed to result from impaired coordination between the detrusor and sphincter functions and reflects a lesion of the spinal cord between the pontine-mesencephalic center regulating the vesicourethral unit and the sacral area. Bladder sensation is intact in some children. This latter type of upper motor neuron defect results in a high incidence of bladder trabeculation, an elevated resting bladder pressure, and dilatation of the upper urinary tract, often reaching enormous proportions (414). Continence appears to depend more on preservation of detrusor activity than of sphincter function. Serial neurologic evaluations indicate that these abnormalities are not static, but tend to change, particularly during the first year of life. Some children with complete or nearly complete denervation of the external sphincter improve, whereas others deteriorate (415). When deterioration occurs in childhood, it most likely occurs in patients with dyssynergia with a small, trabeculated, noncompliant bladder (416). Persistent bacteriuria is seen in 50% of 2-year-old children, with hydronephrosis being found in 25% (417).
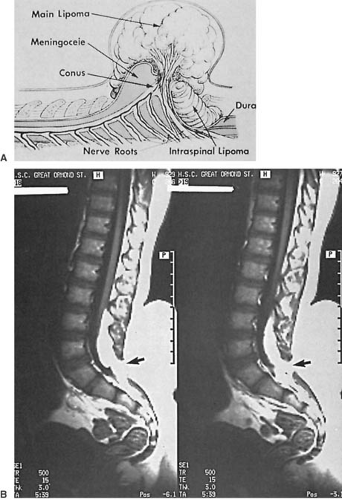 FIGURE 5.4. A: Diagrammatic sagittal view of a lumbosacral lipomyelomeningocele. The subcutaneous lipoma extends through the defect in the posterior arches to end in the low-lying conus medullaris. The skin over the lesion is fully epithelialized, and it had been covered by a large tuft of hair. (From Milhorat TH. Pediatric neurosurgery. Philadelphia: Davis, 1978. With permission.) B: Lipomyelomeningocele. T1-weighted magnetic resonance imaging shows the subcutaneous lipoma extending through the defect in the posterior arches (arrow) into the low-lying conus. (Courtesy of Dr. Brian Kendall, Institute of Neurology, London.) |
The three fundamental urologic problems are infection, incontinence, and retrograde high pressure on the upper urinary tract, producing hydronephrosis and hydroureter. Therefore, early and constant monitoring of the urinary tract with intravenous pyelograms, cultures with colony counts, and voiding cystography is an essential part of any therapeutic program (418). To assess the efficacy of clean intermittent catheterization and to time appropriate surgical intervention for the prevention or arrest of upper urinary tract damage, more complex urodynamic studies are available.
Spina Bifida Occulta.
Spina bifida occulta, referring to a simple bony anomaly in which there has not been complete fusion of the laminae in the midline, is extremely
common. It is found in 25% of children hospitalized for any reason and in 10% of the general pediatric population. It generally involves the posterior arches of L5 and S1. Although it is usually asymptomatic and is found incidentally on radiographic examination, the skin of the low midback region can manifest a hairy tuft, dimple, dermal sinus, or mass caused by a subcutaneous lipoma or teratoma. In the child who has a neurogenic bladder; foot deformities, particularly a broad, shortened, or elevated arch of the foot; or a variety of neurologic deficits of the lower limbs, spina bifida occulta can suggest an underlying malformation of the spinal cord (383,391). In these patients, neurologic deficits, even in the absence of urinary tract or cutaneous abnormalities, are an indication for neuroimaging studies.
common. It is found in 25% of children hospitalized for any reason and in 10% of the general pediatric population. It generally involves the posterior arches of L5 and S1. Although it is usually asymptomatic and is found incidentally on radiographic examination, the skin of the low midback region can manifest a hairy tuft, dimple, dermal sinus, or mass caused by a subcutaneous lipoma or teratoma. In the child who has a neurogenic bladder; foot deformities, particularly a broad, shortened, or elevated arch of the foot; or a variety of neurologic deficits of the lower limbs, spina bifida occulta can suggest an underlying malformation of the spinal cord (383,391). In these patients, neurologic deficits, even in the absence of urinary tract or cutaneous abnormalities, are an indication for neuroimaging studies.
Cranium Bifidum Occultum.
The degree of neurologic and developmental damage in this condition depends on the quantity of protruded tissue, the degree of hydrocephalus, and the extent of hindbrain lesions or cerebral hemisphere dysplasias that result from the associated disorder of cellular migration and organization (331,382).
Often, no functional impairment is noted until childhood, by which time mild mental retardation, spastic diplegia, and impaired cognitive function or seizures can be evident. In the newborn, the mass must be distinguished from cephalhematoma, inclusion cysts of the scalp, cystic hygromas, caput succedaneum, and, in the case of anterior defects, nasal polyps. Its location along the midline, with pulsations synchronous with the heart rate, and absence of periosteal new bone formation distinguish cranium bifidum from these other conditions. Skull radiography reveals the bony defect, and neuroimaging studies define the ventricular system and quantity of neural tissue within the sac.
Treatment
Spina Bifida Cystica.
The management of spina bifida cystica was given new energy by English orthopedic surgeons in the early 1960s (419,420). Their studies suggested that skin closure within 24 hours of birth reduces mortality and morbidity from meningitis and ventriculitis. They argued that early closure not only prevents local infection and trauma to the exposed neural tissue, but also avoids stretching additional nerve roots, which is likely to occur as the cystic sac expands during the first 24 hours. As a consequence, further deterioration of lower limb function and sphincter control is prevented, and motor power of the legs is maintained (421).
In 1971, Lorber (422) proposed the principle of selective surgery and suggested four adverse criteria: a high level of paraplegia, clinically evident hydrocephalus present at birth, congenital lumbar kyphosis, and other major malformations. Other workers, however, have obtained a relatively good outcome in approximately one-half of infants who would have fared badly according to Lorber criteria (423). A further hindrance to the prediction of the future neurologic status of an infant with myelomeningocele is the subsequent progressive cavitation of the cervical and thoracic spinal cord that produces increasing weakness and spasticity of the upper extremities and causes progressive scoliosis (424).
The questions as to whether and when to operate on neonates with spina bifida cystica have perhaps generated more concern and anxiety than any others in pediatric neurosurgery. Many clinicians have refrained from operating on children who had one or more of Lorber adverse criteria. In a paper published in 1974, these selected infants were found to have a 2-year survival rate of 0% to 4% (425).
The reluctance of many American clinicians to follow Lorber criteria in carefully selecting children to treat surgically has been justified by the observation that two or more of the contraindications to operation are commonly compatible with survival and with a quality of life more acceptable than had been expected. In addition, with modern methods of management the purported selection criteria advocated in the past have been shown to have little prognostic value, and in terms of mortality, the ultimate outcome for patients in unselected series compares favorably with that of patients managed according to selection criteria. The effect of early treatment on disability is far less, however. As a rule, the higher the sensory level, the lower is the survival rate, the lower is the IQ, and the lower is the likelihood of employability (426). For a review of the present position, see the papers by McLone (427) and Hobbins (428). A survey published in 1990 found that in an unselected adult population of myelomeningocele patients, some 50% of survivors were ambulatory and 25% were continent. Of the survivors, 50% were able to live without supervision in adapted accommodations and 70% were employable, 25% being able to manage competitive employment (426). McLone concluded that “nearly all children born with a meningomyelocele should have the lesion repaired surgically within 24 hours of birth and should have hydrocephalus treated by shunt diversion … and other appropriate management” (429).
Almost all workers agree that when a patient has been selected for surgical treatment, the procedure should be undertaken within 24 hours of birth and no later than 1 week of age. The claim that delivery by cesarean section before the onset of labor results in better motor function requires confirmation (430), as do the benefits of an in utero repair of the myelomeningocele (431). Early surgery undoubtedly prevents further loss of functioning neural tissue as a result of trauma and infection. Additionally, prompt closure results in shorter hospitalization, easier care of the infant, and psychological benefit to the family and nursing staff. It is important to emphasize to the family that closing the defect does not reverse the neurologic
impairment already present, and that often much additional treatment will be necessary. It is our view that if major malformations of other organ systems are present or if MRI demonstrates major abnormalities of cortical architecture, parents should be advised of these malformations and of the likelihood of a poor intellectual and functional outcome. As a rule, intelligence is related to the thickness of the cortex at the time of shunt insertion and to the sensory level present at birth, with infants who had a lesion in the thoracic region faring worse than those who had a lumbar or sacral lesion (432,433). Children who required a shunt because of hydrocephalus did not perform as well intellectually as those who did not. In particular, those children whose shunt required one or more revisions showed a significant reduction in their cognitive score (433). Bier and colleagues also stressed the importance of socioeconomic factors in the ultimate outcome, particularly in the verbal scores of affected children (433).
impairment already present, and that often much additional treatment will be necessary. It is our view that if major malformations of other organ systems are present or if MRI demonstrates major abnormalities of cortical architecture, parents should be advised of these malformations and of the likelihood of a poor intellectual and functional outcome. As a rule, intelligence is related to the thickness of the cortex at the time of shunt insertion and to the sensory level present at birth, with infants who had a lesion in the thoracic region faring worse than those who had a lumbar or sacral lesion (432,433). Children who required a shunt because of hydrocephalus did not perform as well intellectually as those who did not. In particular, those children whose shunt required one or more revisions showed a significant reduction in their cognitive score (433). Bier and colleagues also stressed the importance of socioeconomic factors in the ultimate outcome, particularly in the verbal scores of affected children (433).
If the neonate is to be treated, as is usually the case, the sac is kept clean and moist before surgery by an undercover of gauze sponges wet with a povidone-iodine solution. To prevent colonization of the gastrointestinal tract and to keep the meconium sterile, the infant is not fed. Systemic broad-spectrum antibiotics, especially against Staphylococcus and coliform organisms, are started when the infant arrives at the hospital and are continued for several days after closure of the sac. Postoperatively, the infant is kept prone for the first week to diminish the risk of urine or feces contaminating the wound. If fluid is present beneath the skin at the repair site, it can be aspirated and a pressure dressing can be applied. Persistent fluid buildup indicates an accumulation of CSF at this location and requires the insertion of a ventriculoperitoneal shunt, even though the ventricles can still be only mildly dilated. Preventing the accumulation of CSF at the repair site allows the wound to heal completely, and rarely is any additional surgery needed at this site. Bladder emptying is assured either with Credé maneuver or with intermittent catheterization. If Credé maneuver is used, occasionally catheterizing the infant is advisable to confirm that the residuals are low.
In the course of follow-up examinations, the head circumference and the appearance of the fontanelle are monitored, and imaging is used whenever the findings suggest increased intracranial pressure. As judged by imaging studies, more than 90% of infants with spina bifida cystica ultimately develop progressive hydrocephalus. Of these, 80% do so within the first 6 months of life and require a shunting procedure (434,435). Approximately 20% of children with myelomeningocele develop symptoms of hindbrain, cranial nerve, or spinal cord compression (436,437). In the majority of cases, manifestations develop before the age of 3 months (408). If the infant develops progressive lower cranial nerve palsies and brainstem signs, it often becomes necessary to decompress the posterior fossa and upper cervical spine, assuming that the hydrocephalus is under good control (437).
Contracture deformities of the lower limbs require physical therapy, leg braces, and stabilization of dislocated hips. Muscle or tendon transplants and joint arthrodeses might be necessary in the ambulating child. Postoperatively, neuropathic fractures resulting from paralysis and prolonged immobility are common. They are best prevented by early active and passive range-of-motion exercises (438).
Kyphosis is an occasional serious and sometimes life-threatening development as the child assumes the sitting posture. It occurs in the more severe cases of extensive lumbosacral myelomeningocele as a result of the paralysis of trunk muscles and from the bone deformity associated with the primary lesion. The spinal deformity poses a threat to respiratory function, to the health of the skin overlying the repaired lesion, and sometimes to the function of surviving cord and nerves. An early decision with respect to reducing the deformity must be made in these circumstances. The deformity is best reduced by the difficult procedure of excision of the vertebral bodies at the level of the kyphosis. In some severe cases, it is possible to perform bony excision at the time of the primary operation. Not providing such treatment can cause a marked reduction in life expectancy.
Spinal cord tethering, of the type found in spina bifida occulta, is present in a small proportion of patients with myelomeningocele. This tethering occurs in addition to the obvious union of the neural elements with the superficial tissues, which is the essential feature of a myelomeningocele. Thus, diastematomyelia, other forms of tethering, and hydromyelia can be present. These additional lesions can be detected by MRI. In the series of Caldarelli and colleagues, routine MRI screening disclosed cavitation of the spinal cord in 22.5% of spina bifida patients. Approximately one-half of the lesions were clinically asymptomatic (439). Deciding whether and when to intervene surgically when such a lesion is found is difficult and requires considerable neurosurgical judgment.
Disorders of the excretory system are the most common cause of morbidity and mortality in patients who survive longer than 2 years. Fernandez and colleagues outlined five points that are invaluable in the management of the child with neurogenic bladder. These are (a) achieving urinary continence, (b) achieving good bladder emptying, (c) lowering intravesical pressure, (d) preventing urinary tract infections, and (e) treating vesicoureteral reflux (440).
In lumbosacral spina bifida cystica, few children attain urinary continence, although McLone and his group believe that bladder and bowel control can be achieved by school age in almost 90% of surviving children (429). The mechanism for urinary incontinence cannot be predicted by the neurologic examination; instead it requires urodynamic testing, including cystometrography, uroflometry,
and electromyography of the urinary sphincter (441). When these studies show the bladder to be atonic but with adequate urethral resistance, treatment is by intermittent catheterization, often in conjunction with cholinergic agents such as bethanechol, which can reduce the residual volume of the bladder. When the bladder is atonic and urethral resistance is inadequate, treatment should be directed to increasing outlet resistance. One way this can be accomplished is by creating an artificial urinary sphincter. Ephedrine, an α-adrenergic agent that acts on the bladder neck, or imipramine can improve continence in the denervated bladder by increasing muscle tone, thereby increasing resistance to bladder outflow. Conversely, this resistance can be diminished by phenoxybenzamine or diazepam.
and electromyography of the urinary sphincter (441). When these studies show the bladder to be atonic but with adequate urethral resistance, treatment is by intermittent catheterization, often in conjunction with cholinergic agents such as bethanechol, which can reduce the residual volume of the bladder. When the bladder is atonic and urethral resistance is inadequate, treatment should be directed to increasing outlet resistance. One way this can be accomplished is by creating an artificial urinary sphincter. Ephedrine, an α-adrenergic agent that acts on the bladder neck, or imipramine can improve continence in the denervated bladder by increasing muscle tone, thereby increasing resistance to bladder outflow. Conversely, this resistance can be diminished by phenoxybenzamine or diazepam.
When incontinence results from a spastic bladder and decreased bladder capacity and urethral resistance is adequate, the high intravesical pressure is managed with anticholinergic drugs. For this purpose, Fernandez and colleagues recommend oxybutynin chloride (0.2 mg/kg per day given in two divided doses), which inhibits the muscarinic action of acetylcholine on smooth muscle (440). Other drugs that have been suggested include propantheline bromide and imipramine hydrochloride. Serial cystometrograms are indicated to monitor drug response. When intermittent catheterization and anticholinergic drugs are insufficient, urinary diversion or bladder augmentation should be considered, with the latter procedure being used more frequently in children (440).
When incontinence is of mixed origin, a combination of medication, intermittent catheterization, and implantation of an artificial urinary sphincter should be tried. In all instances, however, an associated malformation of the urinary tract, such as double ureter or single kidney, must be kept in mind and must be diagnosed to provide comprehensive care.
In the absence of vesicoureteral reflux and in the presence of normal upper tracts, urine cultures taken every 6 months and urograms or imaging studies done every 13 years should suffice. Acute urinary tract infection demands prompt and appropriate antibiotics because infection inevitably leads to persistent vesicoureteral reflux. Such reflux, together with residual urine, produces trigonal hypertrophy and results in retrograde high pressure and eventual hydronephrosis.
Vesicoureteral reflux should be monitored by isotope cystography and requires long-term treatment with low doses of antibiotics such as nitrofurantoin or trimethoprim sulfa. Urinary acidification can be useful in inhibiting calculus formation.
Bowel incontinence owing to a flaccid external sphincter, although not as serious a medical problem as bladder incontinence, poses a much greater social disability. Constipation and impaction of stool are major problems after the first few years of life. Routine enemas or suppositories and biofeedback training have been used with some degree of success, especially when anorectal manometric data demonstrate some rectal sensation and when the patient is able to effect some contraction of skeletal muscle (442,443). Another approach to alleviating the fecal incontinence can be achieved in carefully selected and motivated children who have failed to respond to the other approach and wish to avoid a colostomy. By means of an appendicostomy, a relatively simple operation, the appendix is inserted into the anterior aspect of the cecum, and retrograde colonic enemas can be performed. The slow washing out of the colonic contents by injection of water through the appendicostomy may be needed only every 24 to 48 hours, leaving the child free to take part in normal activities for the remainder of the time. The procedure can be carried out by itself or in combination with surgery for urinary incontinence (444).
The long-term care of the patient with spina bifida cystica requires a multidisciplinary effort. In addition to continuing neurologic and neurosurgical evaluations, the infant also should be seen at regular intervals by orthopedic surgeons, urologists, physiotherapists, and nursing and social services. Details of ongoing care are covered more extensively in a multiauthored book edited by Rekate (445).
Spina Bifida Occulta.
The availability of neuroimaging has allowed a more complete diagnosis of occult dysraphism. Diagnosis is particularly important because the lesions are frequently multiple and surgical intervention at more than one level is indicated.
Clear indications for surgery include the finding of progressive neurologic defects, the presence of an associated tumor or a dermal sinus that carries the risk of meningitis or deep abscess, or a history of meningitis. Considerable debate exists about the need to operate when a malformation has been discovered in the absence of a progressive neural loss. It is our opinion that a child with a lower limb malformation (a small and deformed foot is the most common manifestation) or with a skin stigmata should have plain radiography of the entire spine or MRI studies whenever these are available. When vertebral column malformation is disclosed on the radiographic films, follow-up imaging studies are necessary. Should these reveal an abnormal attachment of the cord or nerve roots, operative intervention to remove that attachment is required as a prophylactic measure, even when no history of progressive damage exists.
Meningocele.
If the meningocele is fully epithelialized and not draining CSF and if the skin over the sac is not ulcerated, immediate repair is not needed and surgery can be deferred for several months. As is the case with spina bifida cystica, the possibility of an additional dysraphic anomaly such as a tethered cord or diastematomyelia requires complete imaging studies. Lesions that protrude
minimally do not necessarily require surgical intervention. Imaging studies of the brain are suggested to assess ventricular size and to detect those few infants who develop hydrocephalus. A small proportion of meningoceles present ventrally as a pelvic mass, or, even more uncommonly, as a posterior mediastinal mass. These lesions are usually not detected in the newborn period.
minimally do not necessarily require surgical intervention. Imaging studies of the brain are suggested to assess ventricular size and to detect those few infants who develop hydrocephalus. A small proportion of meningoceles present ventrally as a pelvic mass, or, even more uncommonly, as a posterior mediastinal mass. These lesions are usually not detected in the newborn period.
Cranium Bifidum Cysticum.
Cranium bifidum cysticum requires immediate repair if CSF is leaking or if the defect is not covered by skin. If the defect is completely epithelialized, it should be closed before the infant’s discharge from the hospital; if the lesion is small and less unsightly, closure can be postponed until later in the first year of life. When the lesion is tender and a source of distress to the infant, early surgical repair is indicated. Posterior encephaloceles are often associated with posterior fossa malformations leading to hydrocephalus. MRI should be done before surgical operation in all cases. Anterior encephaloceles should be repaired by a neurosurgeon working with a cosmetic surgeon.
Prognosis
Follow-up studies have been performed to compare the survival and quality of life among patients treated without surgery, patients who were operated on selectively (i.e., if they lacked the adverse criteria), and patients who received routine early operation (446). Some 45% of infants with myelomeningoceles who are not treated surgically die within the first year of life, most often as a consequence of hydrocephalus or CNS infections (447). Of the survivors, approximately 50% are minimally handicapped (448). The rest are severely handicapped. With operation, approximately 90% survive into their teens, but less than 33% of these are minimally handicapped (449).
Nonselective surgical intervention results in a large number of survivors with major disabilities. By adolescence, 66% of this group are wheelchair dependent because many will have given up assisted walking as they gained weight. Furthermore, 40% have IQs below 80; 66% have no continence of bladder and bowel; 66% have visual defects, including strabismus, corneal scarring, and blindness; 25% have a seizure disorder; and 25% develop precocious puberty. Approximately 90% experience pressure sores, burns, and fractures (450). Incontinence becomes the dominant issue in the surviving adolescent; some undergo urinary diversion and others respond to intermittent catheterization. In most cases, incontinence adversely affects their academic and social life, and more than 90% can be expected to have some degree of personal, social, and economic dependence (451). Long-term studies on patients who had selective surgical intervention have not been completed.
When death occurs after early childhood, it is usually the result of urinary tract infection with sepsis and renal failure. Less often, it is the consequence of increased intracranial pressure resulting from poorly treated or intractable hydrocephalus. In some cases, death is the consequence of pulmonary disease caused by progressive kyphoscoliosis.
Even though surgical and medical advances have improved the prognosis for children with spina bifida cystica, the essence of treatment is prevention of the condition by correction of any maternal nutritional deficiency and prenatal screening programs not only of the high-risk population but also of the general population (452).
Chiari Malformations
The third major expression of dysraphism was first observed by Cleland in 1883 (453), but was more definitively described in Vienna by Chiari in 1891 and 1896 (376,377).1 Chiari malformations are characterized by cerebellar elongation and protrusion through the foramen magnum into the cervical spinal cord. Primary anomalies of the hindbrain and skeletal structures with consequent mechanical deformities produce four different positions of the cerebellum and brainstem relative to the foramen magnum and upper cervical canal (455).
Pathogenesis
Traditional theories of the pathogenesis of the Chiari malformations have been mechanical in nature, but a molecular genetic mechanism of pathogenesis has recently been proposed (456).
The traction theory suggests that tethering of the spinal cord pulls the caudal medulla oblongata and posterior cerebellum through the foramen magnum as the spinal column grows faster than the spinal cord (457). This theory has been totally discredited both experimentally in animals and in humans; traction on the lower spinal cord distorts only the most caudal few segments (458).
The pulsion theory suggests fetal hydrocephalus causes pressure and displacement downward of the brainstem and cerebellum during development (459,460).
The hydrodynamic or oligo-CSF or so-called unified theory of Chiari malformations attributes a paucity of sufficient fluid to distend the cerebral vesicles early in cerebral development because the open neural tube allows leakage and prevents the accumulation of fluid within
the ventricular system (461). Hydrodynamics may indeed play a minor role, but does not explain the entire malformation or its early pathogenesis.
The crowding theory asserts that the posterior fossa itself is too small and the confined neural structures within it are forced through the foramen magnum as they grow.
None of these theories fully accounts for all of the manifestations of Chiari malformations, such as the primary intramedullary dysplasia that usually is found in the brainstem and cerebellum, the accompanying aqueductal stenosis and hydromyelia in many cases, and the inconstant forebrain anomalies in some, such as absence of the corpus callosum. This crowding theory of Marín-Padilla (462) could play a role late in gestation, as the posterior fossa is indeed pathologically reduced in volume, but it cannot explain the pathogenesis of the malformation earlier than 30 weeks, before the cerebellum has grown so that its volume no longer is accommodated in the posterior fossa, and also fails to explain the reason for the small posterior fossa.
The birth trauma theory is wholly without merit, and cases are well documented, both in the literature and in our neuropathologic experience, of fetuses of less than 20 weeks’ gestation with well-formed Chiari malformations (73).
The hypothesis that appears to have the greatest consistency with all aspects of Chiari malformations, particularly types II and III, is a molecular genetic hypothesis that the malformation is caused by ectopic expression of homeobox genes of rhombomere segmentation. The HOX family is implicated in particular because not only do they program segmentation of the hindbrain, but they also are important for the development of the basioccipital, exoccipital, and supraoccipital bones of paraxial mesodermal origin that are hypoplastic in Chiari malformations and result in the abnormally low placement of the torcula and tentorium that causes the posterior fossa to be reduced in volume; HOX genes play no role, by contrast, in the development of membranous bones of most of the cranial vault, which are of neural crest origin (456). Experimentally, Chiari II malformations and often meningomyeloceles may be created in fetal rodents by administering a single dose of retinoic acid (vitamin A) to the maternal animal on embryonic day 9.5 (462). Retinoic acid is known to cause ectopic expression of genes of the Hox and Krox (human EGR2) families in mice, resulting in brainstem and cerebellar malformations. The ependyma shows upregulation of vimentin only in the regions of dysgenesis in Chiari malformations, unlike other cerebral malformations and congenital hydrocephalus of other causes; vimentin expression may be upregulated by genetic mutation of HOX genes (463).
Types of Chiari Malformations
Type I.
In type I malformation, clinically the least severe, the medulla is displaced caudally to the spinal canal and the inferior pole of the cerebellar hemispheres is protruded through the foramen magnum in the form of two parallel, tonguelike processes. This cerebellar displacement can extend as far down as the third cervical vertebra. Though sometimes called a “herniation,” this term is incorrect because it implies that it is being squeezed down, which is not the pathogenesis. Malformations at the base of the skull and the upper cervical spine are sometimes present. These include basilar impression, atlas assimilation, atlantoaxial dislocation, asymmetric small foramen magnum, and Klippel-Feil syndrome. Hydromyelia, syringomyelia, syringobulbia, and diastematomyelia are frequently present (464).
The Chiari malformation type I is generally asymptomatic in childhood, becoming clinically apparent only in adolescence or adult life. The ready availability of MRI studies that demonstrate the malformation has led to a more complete understanding of the clinical spectrum of this condition. Symptoms result from direct medullary compression, compromise of the vasculature supplying the medulla, or, less frequently, from hydrocephalus that develops from aqueductal stenosis or obstruction of the fourth ventricle at its outlet foramina or at the foramen magnum. With obstruction at the foramen magnum, torticollis, opisthotonus, and signs of cervical cord compression are evident. Headache, vertigo, laryngeal paralysis, and progressive cerebellar signs can be accompanied by lower cranial nerve deficits that are often asymmetric (465). Other symptoms of this condition include recurrent apneic attacks and pain in the neck and the occipital region that is exacerbated by laughing or straining. In the series of 43 patients reported by Nohria and Oakes, the mean age at presentation was 17.5 years (466). Hydrosyringomyelia was seen in 65% and scoliosis in 30%. Hydrocephalus was only seen in 12%.
Type II.
In type II, the most common of the Chiari malformations to be diagnosed in childhood, any combination of features of type I malformation can be associated with noncommunicating hydrocephalus and lumbosacral spina bifida. Additionally, the medulla and cerebellum, together with part or all of the fourth ventricle, are displaced into the spinal canal (325,407). The medulla and pons are ventrally linked and juxtaposed (see Figs. 5.2 and 5.3). The pons and cranial nerves are elongated and the cervical roots are compressed and forced to course upward, rather than downward, to exit through their respective foramina. Cervicothoracic hydromyelia and syringomyelia also can be present, and both the foramina of Luschka and Magendie and the basal cisterns are occluded as a result of impaction of the foramen magnum or atresia of the foramina outlets (467).
These anatomic features can be demonstrated readily by MRI studies. Additionally, one can observe that the posterior fossa is smaller than normal and that the tentorium has a low attachment to the occipital bone (407). In 75% of patients, the underdeveloped tentorium allows inferior displacement of the medial posterior cerebrum (444).
In addition to these gross abnormalities, developmental arrests of the cerebellar and brainstem structures, heterotopia of cerebral gray matter, and polymicrogyria also occur. The cortical defects point to an additional defect in cerebral cellular migration and are not explained by obstructive hydrocephalus alone (366,468).
In more than 90% of patients, type II Chiari malformation is seen in conjunction with spina bifida cystica, hydrocephalus, and any combination of the assorted defects already cited for type I (469). Conversely, all patients with spina bifida cystica and hydrocephalus exhibit the type II defect. The clinical presentation of this condition was first noted in 1941 by Adams and colleagues, who demonstrated the lesion by intraspinal injection of lipiodol (470). In the more recent experience of Vandertop and colleagues, 21% of patients develop signs of Chiari II malformation despite good control of their hydrocephalus (437). Symptoms usually develop in infants. They include swallowing difficulties, seen in 71% of the series of Vandertop and coworkers; stridor, seen in 59%; arm weakness, seen in 53%; apneic spells, seen in 29%; and aspiration, seen in 12%. Surgical decompression and sectioning a tight and dense fibrotic band that is often present at the C1 level produces significant improvement in the majority of infants.
Type III.
Type III variant can have any of the features of types I or II. Additionally, the entire cerebellum is herniated through the foramen magnum with a cervical spina bifida cystica. Hydrocephalus is seen regularly and is the result of differing degrees of atresia of the fourth ventricle foramina, aqueductal stenosis, or impaction at the foramen magnum. Rhombencephalosynapsis can rarely be part of type III (471).
Management
The clinical condition and therapeutic regimen for the various Chiari syndromes are described in other sections (see Spina Bifida Cystica, Cranium Bifidum, and Hydrocephalus).
Other Spinal Cord Dysraphic States
The vast majority of other dysraphic states are confined to the spinal cord and its vertebral and cutaneous environment. Like anencephaly and spina bifida, they result from a combination of environmental and genetic components (472). They are of subtle expression and usually are associated with spina bifida occulta along with a tethered cord, a mass lesion, or both (473). A tethered conus medullaris is diagnosed by its position below L2 and by a filum terminale that is wider than 2 mm and is located in the posterior portion of the spinal cord. Tethering results in traction and damage to neural tissue. If a mass lesion is present (e.g., lipoma, syrinx), compression of neural tissue occurs (473).
Neurologic dysfunction can be present already in the neonate with an occult dysraphic lesion. If so, it most frequently involves the lower lumbar and sacral segments and produces motor and sensory loss or sphincter impairment. Musculoskeletal deformities primarily involve the foot, but scoliosis can develop. With somatic growth, the neurologic deficit can worsen or can become apparent if not already present. The progression and development of neurologic symptoms is believed to result from the differential growth between the spinal column and the spinal cord, compression of neural elements, or damage from repeated local trauma (397). Normally, the conus medullaris ascends from the lower lumbar to the upper lumbar level during growth. If tethered by a lipomatous mass, a hypertrophic filum terminale, or a bony spur such as is associated with diastematomyelia, spinal cord ascent is impaired, causing neural damage.
Dysraphisms are often heralded by cutaneous or subcutaneous lesions, such as a hairy tuft, hemangioma, lipoma, sinus, or dimple. We advise imaging studies of the spine and the spinal cord in any neonate with these cutaneous abnormalities. If a patient has neurologic abnormalities and a bony defect in addition to the cutaneous lesions, imaging and surgical exploration are indicated.
In some cases, successful intrauterine surgery of the fetus to repair the meningomyelocele has resulted in dramatic correction of anatomic defects including hydrocephalus and lessening or correction of the Chiari malformation, and this evidence has been cited in support of the hydrodynamic theory of pathogenesis (474,475), but the relief of some anatomic defects to lessen clinical manifestations does not detract from a molecular theory of pathogenesis.
Diplomyelia and Diastematomyelia
Diplomyelia is a complete duplication of a region of spinal cord; diastematomyelia is a vertical division of the spinal cord into two separate halves, usually by an abnormal bony, cartilaginous or fibrous septum over several segments. These two conditions are traditionally discussed together, but they actually are quite different with distinct mechanisms of formation, though not all of the details of pathogenesis are known.
Based on neuroimaging, Pang and coworkers used the term split cord malformation (SCM) and distinguished two types (476), but both really correspond to diastematomyelia. In the first, each of two hemicords is each contained within its own dural tubes and they are separated by a rigid osseocartilaginous median septum. This condition generally corresponds to diastematomyelia. In the second,
the two hemicords are housed in a single dural tube and are separated by a nonrigid fibrous median septum. The state of the dural tube and the nature of the median septum can be demonstrated by imaging studies (476). For this purpose, high-resolution, thin-cut, axial computed tomographic (CT) myelography using bone algorithms is more sensitive than MRI (477).
the two hemicords are housed in a single dural tube and are separated by a nonrigid fibrous median septum. The state of the dural tube and the nature of the median septum can be demonstrated by imaging studies (476). For this purpose, high-resolution, thin-cut, axial computed tomographic (CT) myelography using bone algorithms is more sensitive than MRI (477).
According to Pang and colleagues, both types of SCM result from adhesions between ectoderm and endoderm, which lead to an accessory neuroenteric canal around which an endomesenchymal tract condenses, which bisects the developing notochord and causes the formation of two hemineural plates (476). A true vertical bisection of the notochord would yield two complete spinal cords, not two hemicords, because two floor plates would be induced, though the resulting two spinal cords may have imperfect architecture (73).
Diplomyelia represents a complete duplication of the spinal cord, usually in the lumbar region, 79% in the series of Pang, in which they mistakenly assumed that diplomyelia was a form of diastematomyelia, and occasionally extending for 10 segments or more (477). Because it is a true duplication, each affected spinal segment has four dorsal roots and four anterior horns. The lesion can be associated with extensive spina bifida cystica or with tumors of the spinal cord or it can occur in the absence of other neurologic lesions (478). Diplomyelia could be caused by upregulation of an early gene, expressed in the primitive streak stage, that could also cause conjoined twinning in animals, such as Wnt-8c, or diplomyelia could result from upregulation in the caudal part of the neural tube of a ventralizing gene from the notochord, such as SHH, that causes duplication of the neuraxis; defective SHH expression conversely causes sacral agenesis with severe dysplasia of the spinal cord in those segments (479).
The term diastematomyelia is derived from the Greek diastema, meaning cleft. The condition is marked by a cleft in the spinal cord, which becomes divided longitudinally by a septum of bone and cartilage emanating from the posterior vertebral arch and extending anteriorly. Each half of the cord has its own dural covering. The cord or cauda equina becomes impaled by the bony spur, and differential growth between vertebral column and spinal cord results in stretching of the cord above its point of fixation. An alternative, pathogenetic hypothesis postulates that progressive cord or cauda equina damage results from minor trauma and traction at the spur during head and neck flexion in the growing child. In the majority of patients, diastematomyelia is confined to the low thoracolumbar region, usually extending for 1 to 2 segments, rarely for as many as 10 segments (480). Diplomyelia and diastematomyelia no longer should be regarded as variants of the same process because the pathogenesis is different and the neuropathologic lesions are certainly quite different.
Some 80% to 95% of patients with diastematomyelia are female (481). A variety of skin lesions mark the site of the defect. Most commonly [56% of cases in the series of Pang (477)], these are tufts of hair or dimples, but subcutaneous lipomas, vascular malformations, or dermal sinuses also can be present. Additionally, anomalies of the craniobasal bones may occur as well as syringomyelia and hydromyelia (see Fig. 5.1C). A neurenteric cyst (a persistent embryonic communication with the gut) can be located in the cleft portions of the spinal cord (482).
Progressive sensorimotor deficits represent the most common clinical manifestation for both diplomyelia and diastematomyelia (477,483). They were seen in 87% of symptomatic children and are commonly asymmetric. Pain was seen in 37% of children. Sensorimotor deficits may take two forms. The first is a predominantly unilateral, nonprogressive hypotonia and weakness. The second syndrome is seen in approximately two-thirds of cases. In this entity, the patient experiences weakness and spasticity of the lower extremities with awkward gait, incontinence of bladder and rectum, and, less commonly, posterior root pain. Symptoms either appear de novo or are superimposed on the first syndrome. A combination of upper and lower motor neuron signs in the lower extremities is associated with atrophy of the leg muscles and skeletal deformities of the feet (484). The difference in shape and dimensions of the lower limbs is often quite striking and is believed to result from a combination of prenatal and postnatal asymmetry of growth stimulus secondary to differences in nerve supply. Spinothalamic and posterior column sensory deficits correspond with the level of the lesion. With suspicion aroused by cutaneous anomalies and neurologic dysfunction of the lower limbs and sphincters, diagnosis can best be made by means of MRI of the spinal cord. Because sagittal images may be difficult to interpret in patients with severe scoliosis, coronal images should always be obtained.
Surgery for SCM type I cases (diastematomyelia) has a higher risk of morbidity than is seen for SCM type II cases (diplomyelia), particularly when one hemicord is markedly smaller and, therefore, more delicate (477). Surgical removal of the bony spur allows the cord to become freely movable. Although this does not alter the nonprogressive hypoplastic syndrome, it prevents the onset or arrests and even improves the progressive de novo syndrome (482,485). The more recent the neurologic deficit, the more likely it is to be reversible; hence, prophylactic surgery for the infant or young child without neurologic deficit is indicated. Periodic postoperative follow-up is necessary because regrowth of bone spurs has been reported (486).
Syringomyelia and Hydromyelia
Hydromyelia is an enlargement or dilatation of the central canal of the spinal cord. Syringomyelia involves extension of an enlarged central canal into the cord
parenchyma, usually one or both dorsal horns and dorsal columns. Syringomyelia and hydromyelia are traditionally believed to represent different expressions of the same pathologic process because hydromyelia at times can progress to syringomyelia. However, hydromyelia can occur as a developmental anomaly, associated with Chiari malformations in particular, whereas syringomyelia can be an acquired lesion of the spinal cord secondary to trauma, infarction, or intramedullary tumors. The two terms thus are not synonymous and interchangeable.
parenchyma, usually one or both dorsal horns and dorsal columns. Syringomyelia and hydromyelia are traditionally believed to represent different expressions of the same pathologic process because hydromyelia at times can progress to syringomyelia. However, hydromyelia can occur as a developmental anomaly, associated with Chiari malformations in particular, whereas syringomyelia can be an acquired lesion of the spinal cord secondary to trauma, infarction, or intramedullary tumors. The two terms thus are not synonymous and interchangeable.
As originally defined, syringomyelia is a condition of tubular cavitation within the spinal cord. When the cystic lesion extends into the medulla and pons, the condition is termed syringobulbia. When hydromyelia is a developmental defect from early fetal life, the large central canal is fully lined by ependyma, though it overexpresses vimentin (463). By contrast, the hydromyelia that results from acquired injuries postnatally usually leaves a central canal with ependymal discontinuities and large gaps.
When syringomyelia is associated with Chiari type I or II malformation, as it often is, it is accompanied by tonsillar herniation and an apparent occlusion of the outlet foramina of the fourth ventricle. The diameter of the cervical spinal canal is enlarged, and extensive scoliosis or kyphoscoliosis can be present. Some cases of syringomyelia are post-traumatic, a condition more fully covered in Chapter 9 (Fig. 5.5). Others, nearly 20%, are associated with a spinal cord tumor (487). Rarely, spinal cord gray and white matter is disorganized, a microscopic picture analogous to schizencephaly of the cerebral cortex. In the majority of cases, however, the cause of syringomyelia remains essentially unknown despite numerous theories and many clinical and experimental studies intended to explain the condition (488). One theory, as proposed by Oldfield and colleagues, is based on their observations, supported by MRI and cine-MRI, that in patients with syringomyelia associated with the Chiari I malformation the subarachnoid space is partially occluded at the level of the foramen magnum by the displaced brainstem and cerebellar tonsils (489). This displacement impedes the rapid upward and downward movement of CSF during the cardiac cycle and produces a systolic pressure wave in the spinal CSF that forces CSF into the spinal cord through the perivascular and interstitial spaces.
Hydromyelia, a dilated central canal without extension into the spinal cord parenchyma, resembles the healthy fetal condition at midgestation, although often in a more extreme form. At 6 to 12 weeks’ gestation, the central canal is a tall, vertical slit extending almost the entire vertical axis of the neural tube. It is initially lined by undifferentiated neuroepithelium with mitotic activity at the surface of the central canal; a pseudostratified columnar ependymal epithelium then develops and stops all mitotic activity, first at the basal plate and later at the alar plate. The slit becomes round in transverse sectional views and progressively narrows as the fetus and infant mature. In early adult life the canal ceases to exist altogether and its site is identified by clusters and rosettes of residual ependymal cells, but the lumen is obliterated. Hydromyelia of the cervical and often of the thoracic and lumbosacral spinal cord usually accompanies Chiari II malformations, and the ependyma lining it overexpresses vimentin as a persistent fetal feature that normally disappears by 34 weeks’ gestation. Vimentin expression continues into adult life in patients with Chiari malformations, but does so only in the regions of dysgenesis of the ventricles, not in the normal lateral or third ventricles or in normal segments of the spinal cord (463).
Though sometimes recognized during childhood, syringomyelia usually does not become symptomatic until adult life (489). The vast majority of lesions involve the
cervical cord, and, therefore, the upper extremities are preferentially involved. However, a syrinx is sometimes encountered in spinal dysraphism of the lower spine, when it is usually associated with other malformations in this region. Because the cavities tend to be more central than eccentric, they damage fibers crossing through the central white matter of the cord and thus compromise temperature and pain sensation, sparing sensory modalities mediated by the posterior columns. This disassociated anesthesia is responsible for cutaneous trophic, sudomotor, and vasomotor disorders, including painless ulcerations, coldness, cyanosis, and hyperhidrosis. It also causes the painless arthropathies, the Charcot joints similar to those seen in tabes dorsalis, but involving joints of the upper rather than the lower extremities.
cervical cord, and, therefore, the upper extremities are preferentially involved. However, a syrinx is sometimes encountered in spinal dysraphism of the lower spine, when it is usually associated with other malformations in this region. Because the cavities tend to be more central than eccentric, they damage fibers crossing through the central white matter of the cord and thus compromise temperature and pain sensation, sparing sensory modalities mediated by the posterior columns. This disassociated anesthesia is responsible for cutaneous trophic, sudomotor, and vasomotor disorders, including painless ulcerations, coldness, cyanosis, and hyperhidrosis. It also causes the painless arthropathies, the Charcot joints similar to those seen in tabes dorsalis, but involving joints of the upper rather than the lower extremities.
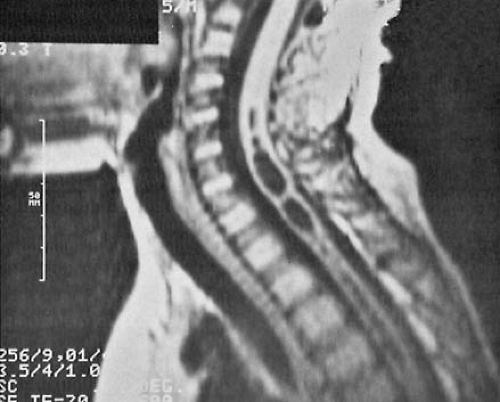 FIGURE 5.5. Cervical syringomyelia. Magnetic resonance imaging study. The syrinx was essentially asymptomatic in this 4-year-old boy. It was discovered when imaging studies were done as part of an evaluation for a hemiplegia, which was found to be caused by a parietal porencephaly. |
As the syrinx enlarges, involvement of anterior horn cells, pyramidal tracts, and posterior columns in the cervical region leads to segmental asymmetric lower motor neuron signs in the upper and lower extremities. Spasticity, hyper-reflexia, and loss of position and vibratory sense occur. In children, the first sign can be a rapidly progressive scoliosis (490). In syringobulbia, asymmetric lower cranial nerve palsies occur, and the condition is almost always associated with anomalies of the craniobasal bones. Brainstem tumors are occasionally present. Syrinxes in the child or adult can impinge on dorsal motor nuclei of the vagus nerve, producing episodic stridor with laryngospasm, or they can compromise the nucleus ambiguous, causing chronic stridor and vocal cord paralysis (491).
Diagnosis is best made by MRI studies. Sagittal views provide excellent visualization of the cystic cavity and of the anatomy at the level of the foramen magnum. Gadolinium enhancement can be used to verify the presence of an adjoining tumor (492). Considerable dispute exists about the best means of managing the lesion (487,493). Posterior fossa and upper cervical decompression is the recommended treatment for the most common form of syringomyelia seen in the pediatric population (i.e., that associated with Chiari type II malformation and downward displacement of the cerebellar tonsils and outlet obstruction of the fourth ventricle) (487,494). In patients in whom such decompression fails to stabilize the neurologic deterioration and in whom persistence of a dilated cyst can be demonstrated, shunting of the CSF from the cystic cavity into the neighboring subarachnoid space is recommended (487,492). The value of occluding the opening from the fourth ventricle into the syringomyelic cavity has come under question, but is still recommended when syringomyelia is associated with the Dandy-Walker syndrome (487,495,496). Follow-up examinations by MRI are required.
Sacral Agenesis
Sacral agenesis is marked by total absence of the coccyx and the lower two or three sacral vertebrae and hypoplasia of the vertebrae just above the aplastic segments. An associated anomalous development of the lumbosacral cord and other major or minor dysraphisms occur. Lipoma of the conus medullaris and filum terminale often accompany the defect (497). Sacral agenesis is associated in animals and humans with underexpression of Sonic hedgehog at the caudal end of the neural tube. The same molecular defect when expressed only at the rostral end of the neural tube is associated with holoprosencephaly. The gene for sacral agenesis has been mapped to chromosome 7q36, the same region that contains a gene for holoprosencephaly (498). Sacral agenesis, therefore, represents a genetic disorder of neural induction and of failure in induction of the sclerotome in the ventral half of the paired somites. In severe cases, the entire somite may fail to develop at the sacral and lumbar levels, hence the myotome is affected as well and fails to generate myocytes, resulting in segmental amyoplasia. Sensory innervation from the dorsal horns and dorsal roots remains well preserved, by contrast, and autonomic neural structure and function are variably involved (499).
Neurologic signs are those of a flaccid neurogenic bladder with dribbling incontinence, motor deficits, to a lesser extent sensory deficits of the lower extremities, lower extremity muscle hypoplasia, and skeletal arthrogryposis (499). Recurrent urinary tract infection and hydronephrosis, aggravated by delay in diagnosing the primary process, are major sources of morbidity (500). The defect has been associated with imperforate anus, malrotation of the bowel, and genital anomalies. All malformations can be dated to the first 7 weeks of gestation (501). Sacral agenesis is seen in approximately 1% of offspring of diabetic mothers, and Passarge and Lenz postulated that insulin interferes with the differentiation of caudal chordamesoderm (502). The caudal regression syndrome refers to severe sacral agenesis associated with syringomelia or lack of separation of the two legs, as with manatees or seals.
Neurodermal Sinus
The majority of dermal sinuses (e.g., the pilonidal sinus) do not connect with the CNS and are therefore of limited neurologic importance.
Neurodermal sinuses are relatively frequent among cases of occult spinal dysraphism (383). They represent a communication lined by stratified squamous epithelium between skin and any portion of the neuraxis. Most commonly, the defects are in the lumbosacral region and the occiput, defects in the lumbosacral region occurring approximately nine times as frequently as those in the occiput. These two levels are at the terminal closure sites of the neural tube.
The sinus is often surrounded by a small mound of skin, the dimple, or other cutaneous lesions such as tufts of hair or angiomas. It often overlies a spina bifida occulta. It can expand into an epidermoid or dermoid cyst at its proximal end, causing segmental neurologic deficits, either through mass effect or by traction on the neuraxis. Cerebellar and
brainstem signs or, on occasion, hydrocephalus can be produced by a neurodermal sinus in the occipital region. The presence of an open sinus tract can provide a portal of entry for bacterial infections, and a neurodermal sinus is an important cause for recurrent meningitis. In other cases, the dermoid cyst enlarges rapidly through infection of its contents, becoming an intraspinal abscess. When the lesion is in the lumbosacral region, coliform bacteria and staphylococci are the most common invaders; a sinus tract in the occiput is more likely to produce recurrent staphylococcal meningitis. Such defects must be scrupulously sought in any case of CNS infection (503). Any dermal sinus ending that is above the level of the lower sacrum should be traced with neuroimaging studies.
brainstem signs or, on occasion, hydrocephalus can be produced by a neurodermal sinus in the occipital region. The presence of an open sinus tract can provide a portal of entry for bacterial infections, and a neurodermal sinus is an important cause for recurrent meningitis. In other cases, the dermoid cyst enlarges rapidly through infection of its contents, becoming an intraspinal abscess. When the lesion is in the lumbosacral region, coliform bacteria and staphylococci are the most common invaders; a sinus tract in the occiput is more likely to produce recurrent staphylococcal meningitis. Such defects must be scrupulously sought in any case of CNS infection (503). Any dermal sinus ending that is above the level of the lower sacrum should be traced with neuroimaging studies.
These lesions require surgical exploration and complete excision of the sinus before the development of symptoms. An occipital sinus is treated by primary excision of the entire sinus tract, and of the proximal cyst if it is present. In Matson’s experience, surgical results were poorer when performed after the development of infection, with death resulting from chronic meningitis or hydrocephalus (373).
Congenital Scalp Defect
A congenital defect of the scalp, also known as aplasia cutis congenita, is a relatively rare anomaly seen in either sex. It can occur in isolation or in combination with a wide variety of other cerebral or extracranial anomalies (504). Rarely, congenital scalp defects are associated with similar cutaneous lesions elsewhere on the body. In 80% of instances the defect is sporadic; in the remainder it is inherited as autosomal dominant or autosomal recessive traits (331). The defect can vary from one that is small to one that includes most of the calvarial surface. Although in two-thirds of cases the underlying skull is intact, other cases involve underlying defects of periosteum, skull, and dura that often close spontaneously during the first few months after birth. The defect is generally at the vertex, although some lesions occur off the midline. The brain is usually normal, but imaging is justified to verify this assumption. Posterior midline scalp defects can be accompanied by mental retardation, congenital deafness, and hypothyroidism, the Johanson-Blizzard syndrome, and are seen in infants with deletion of the short arm of chromosome 4 (505). A small skin defect can be closed, but if it is large, grafting might be required. It is important to keep the lesion clean and moist before surgical repair. A dermoid or epidermoid cyst may occur in the site of the anterior fontanelle as a benign lesion.
Congenital Defects of the Cranial Bones
Congenital defects of the skull bones without loss of overlying skin or underlying dura result from a failure of ossification that usually occurs at the vertex either as enlarged persistent biparietal foramina or as an absence of the sphenoid wing. The latter can result in a pulsating exophthalmos that can damage the globe or optic nerve. In approximately 50% of the cases, this defect is associated with neurofibromatosis. Diagnosis is made by radiography or CT.
DEVELOPMENTAL ANOMALIES OF THE CEREBELLUM
In the Chiari malformation, the cerebellar hemispheres that remain in the posterior fossa are frequently hypoplastic. On microscopic examination, the cerebellum often shows disorganization of the normal lamination with the Purkinje cells being heterotopic or focally absent.
Complete cerebellar aplasia is a rare condition. It is attributed to destruction of the cerebellum rather than representing a true aplasia (506). However, molecular genetic manipulations in mice demonstrate that global cerebellar aplasia results from homozygous deletion of Wnt-1, En-1, or both genes, and En-2–defective expression results in cerebellar hypoplasia (46). In all three of these genetic mutations, agenesis of the mesencephalon and metencephalon (rostral pons) also is present in the mouse. Each of these genes is essential for the development of the midbrain neuromere (r0) and rhombomere 1 (r1). Nearly identical lesions can occur in humans, and it seems likely that an EN2 mutation accounts for this combination of focal malformations of derivatives of r0 and r1 that cannot be explained as infarction or on any other basis (507).
Cerebellar Hypoplasias
Under cerebellar hypoplasias are grouped several entities in which the cerebellum does not achieve its full developmental potential. Cerebellar hypoplasia can be categorized as global or as selective for the vermis or the lateral hemispheres.
Global hypoplasia of the cerebellum can be caused by a variety of endogenous or exogenous factors. It is seen as an autosomal recessive trait, in a variety of chromosomal disorders, and as a result of intrauterine exposure to drugs or irradiation. Patients with the autosomal recessive condition demonstrate a small cerebellum with an atrophic cerebellar cortex. Granule cells are markedly reduced or absent. Purkinje cells can be normal or can demonstrate a variety of abnormalities. Clinically, children present with delayed development, generalized muscular hypotonia, fixation nystagmus, esotropia, and, in the more severe cases, microcephaly and a seizure disorder (508). Ataxia and intention tremor were seen in all older children reported by Sarnat and Alcalá, but the common presentation in infancy was hypotonia and gross motor developmental delay; phasic nystagmus was variably expressed at any age (509). Migratory disorders of the cerebral cortex frequently accompany this condition (510).
Two syndromes of an autosomal recessive form of pontocerebellar hypoplasia have been reported. One type is accompanied by ventral horn cell degeneration similar to that seen in infantile spinal muscular atrophy; the other is marked by progressive microcephaly and extrapyramidal movements appearing during the first year of life (511). However, pontocerebellar hypoplasias may not be simple developmental malformations, but progresssive degenerative diseases beginning in fetal life, and hence combine hypoplasia with atrophy of incompletely developed structures of the brain and involve supratentorial as well as posterior fossa structures.
Aplasia of the Vermis
Selective aplasia of the vermis can be genetic or acquired. Acquired vermal aplasia or hypoplasia results from a variety of teratogens acting on the brain during the seventh to eighth weeks of gestation, a time when the neural folds of the developing cerebellum meet in the midline to enclose the fourth ventricle and form the vermis (512). Hypoplasia of the cerebellar vermis occurs sporadically, as an autosomal dominant trait (513) or as Joubert syndrome, an autosomal recessive condition. Since its first description in 1969 by Joubert and coworkers (514), this disorder has been reported from all parts of the world. Patients usually present in infancy with respiratory irregularities, notably hyperpnea alternating with apnea. These were seen in 44% of patients in the series of Kendall and colleagues (515). Abnormal eye movements, notably nystagmus and impaired supranuclear control, were seen in 67%. Reduced visual acuity or retinal dystrophy were seen in 44%. Cyclic conjugate lateral deviation of the eyes accompanied by head turning also has been described (516). Hypotonia and mental retardation are common, as are polycystic kidneys and congenital anomalies of the extremities. Variants of Joubert syndrome also are described; in one form, supratentorial white matter shows delayed myelination as a probable autosomal recessive trait in siblings (517). Hypoplasia of the cerebellar vermis also is well documented in many cases of infantile autism, but the genetics is uncertain (518).
Selective aplasia of the vermis may leave a CSF-filled extension of the subarachnoid space that separates the medial borders of the preserved lateral hemispheres, as in Joubert syndrome or associated with Dandy-Walker malformation, or it may be associated with fusion of the medial wall of the hemispheres and of the dentate nuclei that obliterates this space and also causes the fourth ventricle to be shallow, tall, and distorted. This latter condition is termed rhombencephalosynapsis and is most often a cerebellar component of the forebrain malformation septo-optic-pituitary dysplasia, but not of holoprosencephaly (512). The condition occasionally is associated with the Chiari III malformation (471). Children present with cerebellar deficits and mental retardation of variable severity (506,510).
Partial or complete absence of the vermis can be visualized by neuroimaging or on pathologic examination. In some cases the cerebellar folia are normal; in others the hemispheres are hypoplastic with hypomyelination of cerebellar white matter. The brainstem is often hypoplastic with absence of the pyramidal decussations and a variety of abnormalities at the cervicomedullary junction (515). Commonly, an associated atrophy of the cerebral hemispheres occurs. Diagnosis of the condition is best made by MRI. In some cases the secondary enlargement of the cisterna magna bears some resemblance to the Dandy-Walker syndrome (519).
A rare, sporadic malformation in which hypoplasia of the vermis is associated with an occipital encephalocele and ventrolateral dislocation of the hypoplastic cerebellar hemispheres has been termed inverse cerebellum or tectocerebellar dysraphia. Hydrocephalus, microcephaly, agyria, and dysplasia of the corpus callosum are associated findings (520). The Dandy-Walker syndrome, which, in part, is characterized by complete or partial agenesis of the vermis, is covered in another section of this chapter.
Aplasia of the Cerebellar Hemispheres
Selective agenesis of the cerebellar hemispheres with preservation of the vermis is associated with secondary degeneration of cerebellofugal and cerebellopetal tracts, abnormalities in the basal ganglia, microcephaly, and mental retardation (521). Generalized disorganization of the cerebellar cortex usually is accompanied by such major cerebral malformations as holoprosencephaly. Because migration of the external granular layer of the cerebellum continues into the second year of postnatal life, focal dysplasias of the cerebellum can result from either prenatal or perinatal insults. When the dysplasias are prenatal, they are marked by heterotopia of undifferentiated neuroepithelial cells and hypertrophy and proliferation of the cells of the cerebellar cortex.
The condition known as cerebellar hypertrophy, dysplastic gangliocytoma of the cerebellum, or Lhermitte-Duclos syndrome manifests as a cerebellar tumor. It is covered in Chapter 11, although it is really a malformation and not a true neoplasm.
DEVELOPMENTAL ANOMALIES OF THE BASE OF THE SKULL AND UPPER CERVICAL VERTEBRAE
Platybasia
The terms platybasia and basilar impression frequently are used interchangeably, but they are not the same. Platybasia refers to a condition in which the angle formed by a
line connecting the nasion, tuberculum sellae, and anterior margin of the foramen magnum is greater than 143 degrees.2 The diagnosis of platybasia in young infants should be made cautiously because it also represents a normal transitory developmental stage. Basilar impression, by contrast, refers to a pathologic upward displacement of the occipital bone and cervical spine with protrusion of the odontoid process into the foramen magnum. It is a common complication of conditions in which the bones are vulnerable to deformity or easy fracture, as in osteogenesis imperfecta.
line connecting the nasion, tuberculum sellae, and anterior margin of the foramen magnum is greater than 143 degrees.2 The diagnosis of platybasia in young infants should be made cautiously because it also represents a normal transitory developmental stage. Basilar impression, by contrast, refers to a pathologic upward displacement of the occipital bone and cervical spine with protrusion of the odontoid process into the foramen magnum. It is a common complication of conditions in which the bones are vulnerable to deformity or easy fracture, as in osteogenesis imperfecta.
Platybasia also is sometimes applied to a familial disorder characterized by a deformity of the osseous structures at the base of the skull that produces an upward displacement of the floor of the posterior fossa and a narrowing of the foramen magnum. Basilar impression, with which platybasia often is confused, may be transmitted as an autosomal dominant trait with occasional lack of penetrance (522). As a rule, neurologic symptoms do not appear until the second or third decade of life. When they do, they are progressive and are caused by compression of the cervical spinal cord. Commonly, they include progressive spasticity, incoordination, and nystagmus with lower cranial nerve palsies. Platybasia after 2 years of age can be associated with other malformations of the CNS, including the Chiari malformations and aqueductal stenosis.
The diagnosis is suggested by a short neck and a low hairline. It is confirmed by radiography of the skull and upper cervical spine. These reveal an odontoid process that extends above a line drawn from the dorsal lip of the foramen magnum to the dorsal margin of the hard palate.
Treatment is by surgical decompression of the posterior fossa and upper cervical cord (510).
Klippel-Feil Syndrome
Klippel-Feil syndrome, first described in 1912 by Klippel and Feil (523), is characterized by a fusion or reduction in the number of cervical vertebrae. The embryonic defect is a failure of segmentation of the chordamesoderm and its derivative sclerotomes that ultimately go on to form the cervical vertebrae. It is probably a disorder of segmentation of embryonic somitosomes into distinct segmental somites, including the sclerotomes that form the vertebrae. The most likely, though not yet proved, etiology of Klippel-Feil malformation of the cervical spine is defective expression of a HOX-family gene that programs segmentation not only of the hindbrain, but also of somites, including sclerotomes that form vertebral bodies. In most cases the syndrome appears sporadically; in isolated families an autosomal dominant transmission has been recorded.
On examination, affected children have a short neck and a low hairline. Passive and active movements of the neck are limited. Neurologic symptoms are variable. Progressive paraplegia owing to compression of the cervical cord can appear early in life. Some children exhibit retardation or show learning deficits. An association with mirror movements has been reported. It could reflect the soft signs seen in children with mild intellectual deficits, or result from an inadequate decussation of the pyramidal tracts or dorsal closure of the cord (524). Associated malformations are common and include spina bifida, syringomyelia, and fibrosis of the lateral rectus muscle of the eye (Duane syndrome) (Table 5.14). The constellation of Klippel-Feil syndrome, congenital sensorineural hearing loss, and abducens palsy is known as the Wildervanck syndrome (Table 5.15). Wildervanck syndrome is limited to female patients, suggesting that the condition is lethal in the hemizygous male individual. Klippel-Feil syndrome also can be accompanied by congenital heart disease (538,539).
The diagnosis of Klippel-Feil syndrome rests on radiographic demonstration of fused cervical or cervicothoracic vertebrae, hemivertebrae, or atlanto-occipital fusion. MRI can demonstrate compression of the cervical cord, syringomyelia, and other CNS anomalies.
With clinical evidence for compression of the cervical cord, laminectomy is indicated.
Cleidocranial Dysostosis
Cleidocranial dysostosis is transmitted as an autosomal dominant trait and is characterized by rudimentary clavicles, a broad head, delayed or defective closure of the anterior fontanelle, mental deficiency, and a variety of cerebral malformations (540). Other skeletal malformations are common. These include spina bifida, short and wide fingers, and delayed ossification of the pelvis.
ANTERIOR MIDLINE DEFECTS
Holoprosencephaly
Holoprosencephaly is a disorder of forebrain cleavage of the early prosencephalon to form two distinct telencephalic hemispheres. Six different genes have been identified in various patients with holoprosencephaly, yet all six together represent only about 20% of total cases studied, hence there are likely many more yet to be discovered. Of the six known genes, five follow a ventrodorsal gradient or have a ventralizing effect and one (ZIC2) is a dorsalizing gene. A defective expression of the transcription product of the gene Sonic hedgehog (SHH) has been shown to be the responsible molecular event in some cases, both in
animal models and in humans (541,542); defective expression of this gene at the caudal end of the neural tube causes sacral agenesis (see later discussion), but both malformations never occur in the same individual. As a consequence of downregulation of SHH (human locus 7q36qter), the formation of a median (interhemispheric) fissure and the development of paired telencephalic hemispheres from the prosencephalon fail (543). The defect in cleavage continues to influence the development of other cerebral structures that occur at a later time in ontogenesis. For example, the corpus callosum fails to form and, unlike cases of isolated callosal agenesis, a bundle of Probst composed of misdirected callosal axons within the same hemisphere where they arose also is absent. Various migrational abnormalities occur. As with anencephaly, the dysplasia is time specific and the stimulus is nonspecific. Normally, cleavage of the hemispheres occurs at 33 days’ gestation. Thus, of the major induction malformations, holoprosencephaly has the shortest vulnerable period and one of the earliest onsets.
animal models and in humans (541,542); defective expression of this gene at the caudal end of the neural tube causes sacral agenesis (see later discussion), but both malformations never occur in the same individual. As a consequence of downregulation of SHH (human locus 7q36qter), the formation of a median (interhemispheric) fissure and the development of paired telencephalic hemispheres from the prosencephalon fail (543). The defect in cleavage continues to influence the development of other cerebral structures that occur at a later time in ontogenesis. For example, the corpus callosum fails to form and, unlike cases of isolated callosal agenesis, a bundle of Probst composed of misdirected callosal axons within the same hemisphere where they arose also is absent. Various migrational abnormalities occur. As with anencephaly, the dysplasia is time specific and the stimulus is nonspecific. Normally, cleavage of the hemispheres occurs at 33 days’ gestation. Thus, of the major induction malformations, holoprosencephaly has the shortest vulnerable period and one of the earliest onsets.
TABLE 5.14 Unusual Congenital Defects of the Cranial Nerves and Related Structures | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Holoprosencephaly may result from multiple genetic defects. SHH expression can be altered in inborn metabolic diseases of cholesterol synthesis, notably in the Smith-Lemli-Opitz syndrome in which the conversion of the cholesterol precursor 7-dehydrocholesterol to cholesterol is defective. In this condition, which is frequently accompanied by holoprosencephaly, the SHH protein product undergoes autoproteolysis to form a cholesterol-modified active product (544) (see Chapter 1). Other ventralizing genes identified in human holoprosencephaly include SIX3 at 2q21 (545), TGIF at 18p11.3 (546), PTCH, which is an SHH receptor, at 9q22.3 (547), and the head inducer DKK at 10q11.2 (548). In still other cases of holoprosencephaly, a mutation of the zinc-finger dorsalizing gene ZIC2 at 13q32 occurs (549). Because this genetic defect is associated with 13q deletions, it may be the cause of holoprosencephaly in infants with 13 trisomy (550).
Holoprosencephaly generally is sporadic. In Japan, the incidence is 0.4% of aborted fetuses and 6 in 10,000 liveborn babies (550). A variety of chromosomal abnormalities are linked with holoprosencephaly. Besides trisomy of chromosome 13, these include a partial deletion of the long arm of chromosome 13, ring chromosome 13, trisomy of chromosome 18, partial deletion of the short arm of chromosome 18, ring chromosome 18, and partial trisomy of chromosome 7 (551,552) (see Chapter 4). The malformation also is associated with such maternal disorders as diabetes mellitus, syphilis, cytomegalic inclusion disease, and toxoplasmosis (553). The risk of recurrence in siblings of affected patients is 6%. If a chromosomal anomaly is identified, the risk for recurrence is 2%, but is higher if mother is older than 35 years of age (554). After chromosomal disorders, maternal diabetes mellitus, including gestational diabetes, is the most common condition associated with holoprosencephaly.
An autosomal dominant form of holoprosencephaly has been well described. The gene for this disorder has been localized to the telomeric region of the long arm of chromosome 7 (7q36.2) and has been shown to be associated with defective expression of SHH (542,555). In several reported families with this disorder, penetrance has been 88%, with mental retardation representing a mild expression of the defect (556). In a minority of cases, holoprosencephaly has been transmitted as an autosomal recessive condition, and this trait is described in each of the identified genetic mutations (545,546,547,548,549,557,558).
TABLE 5.15 Common Causes for Profound Hearing Loss in Childhood | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Holoprosencephaly has been recognized in various degrees of severity, but these morphologic variants are only degrees of severity and do not refer to the genetic defect because all three anatomic forms have been described in association with each of the six known genetic mutations (559,560,561,562). In its most complete expression (alobar holoprosencephaly), the brain is characterized by a single midline ventricular cavity encompassed within an undivided prosencephalic vesicle (Fig. 5.6). The thalamus remains undivided, the inferior frontal and temporal regions are often absent, and the remainder of the isocortex is rudimentary, with only the primary motor and sensory cortex present. The olfactory bulbs and tracts are absent, and indeed the original descriptive name applied to this malformation was arhinencephaly, but poorly differentiated primordial olfactory bulbs may be adherent to the entorhinal cortex as seen by careful microscopic examination; hence this may be a problem of growth and migration of the olfactory bulbs rather than total agenesis (298). Many other neuropathologic details are well described in holoprosencephaly (298,563,564). The brainstem and cerebellum are present and fully differentiated. Minor focal dysplasias of the cerebellar cortex may be present, however. Gray matter heterotopia and agenesis of the septum
pellucidum result from abnormal cellular migration (565). The lamination and general architecture of the cortex is abnormal, and extensive sites of overmigration occur beyond the limits of the pia mater into the leptomeninges. Some of these overmigrated nodules become isolated from the brain as ectopia and others remain attached to the cerebral surface or connected by a stalk. These nodules are sometimes known by the ignoble term brain warts. In holoprosencephaly, absence or severe deficiency occurs of the external granular layer of Brun, a transient layer of glial cells at the cerebral cortical surface during the stage of radial migrations. Its absence could account for the failure to limit overmigration (298).
pellucidum result from abnormal cellular migration (565). The lamination and general architecture of the cortex is abnormal, and extensive sites of overmigration occur beyond the limits of the pia mater into the leptomeninges. Some of these overmigrated nodules become isolated from the brain as ectopia and others remain attached to the cerebral surface or connected by a stalk. These nodules are sometimes known by the ignoble term brain warts. In holoprosencephaly, absence or severe deficiency occurs of the external granular layer of Brun, a transient layer of glial cells at the cerebral cortical surface during the stage of radial migrations. Its absence could account for the failure to limit overmigration (298).
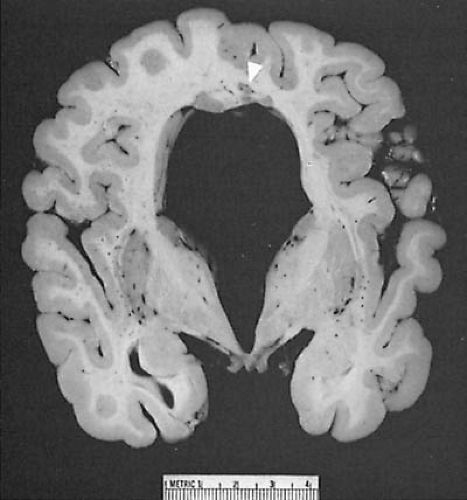 FIGURE 5.6. Holoprosencephaly. In this coronal section, note the common lateral and third ventricles. Midline fusion of the frontal lobes is seen in the absence of the interhemispheric fissure. Note the subependymal heterotopia at the usual location of the corpus callosum (arrowhead). These may give rise to seizure foci. (Courtesy of Dr. Hideo H. Itabashi, Department of Pathology, Los Angeles County Harbor Medical Center, Torrance, CA.) |
One of the most important aspects of brain development in holoprosencephaly is the extent of gradients of genetic expression, regardless of which gene is mutated (298). Most genes affecting the embryonic nervous system have a maximal expression in one part of the neural tube and their effects become lessened or disappear in progressively more distal regions. In holoprosencephaly, the most severe defect in the lateral gradient is in the midline, and more-lateral structures of the cerebral cortex are less affected, so that the most lateral regions of frontal, parietal and temporal and occipital lobes may show histologically normally laminated neocortex, whereas severely disorganized cortical architecture is seen in midline and parasagittal sections, and less severely dysplastic cortex is found in intermediate regions (298). The extent of the mediolateral gradient may determine such clinical expression of cerebral cortical function as epilepsy, degree of mental retardation, and various cognitive defects. The rostrocaudal gradient in the longitudinal axis may extend from the telencephalon to involve the diencephalon, with noncleavage of the thalamus, and may extend further into the midbrain, with noncleavage of the superior colliculi and atresia of the cerebral aqueduct. Involvement of the midbrain neuromere also affects the mesencephalic neural crest formation that determines the extent of the midfacial hypoplasia, and this rostrocaudal gradient is unrelated to the severity of malformation of the forebrain; hence a normal face can be seen in alobar holoprosencephaly if the midbrain is normal, and severe midfacial hypoplasia can occur in the mild lobar form if the midbrain is involved (298).
The clinical picture of this condition is highlighted by midline facial abnormalities, which are seen in the large majority of children with alobar holoprosencephaly and in many cases with milder forms of holoprosencephaly. The clinical and imaging classification of holoprosencephaly into alobar, semilobar, and lobar forms reflects the degree of genetic deficiency and how far caudally in the neural tube the underexpression extends rather than fundamental differences in pathogenesis. In alobar holoprosencephaly, the neurologic picture is characterized by severe to profound mental retardation, seizures, rigidity, apnea, and temperature imbalance. Hydrocephalus can develop as a consequence of aqueductal obstruction, and associated hypothalamic or pituitary malformation can induce endocrine disorders; epilepsy is variable and only a minority of infants with holoprosencephaly have seizures in the neonatal period or later, though infantile spasms and other severe forms of epilepsy can occur (561,562).
The anatomic forms of holoprosencephaly can be defined by CT, MRI, and neuropathologic examination. The most extreme form, alobar holoprosencephaly, is characterized by a monoventricle continuous with the third ventricle, so that foramina of Monro cannot be recognized. This single midline ventricle is large, and the cerebral cortex and hippocampi are continuous across the frontal midline; none of the horns of the lateral ventricles are differentiated. The third ventricle may be atretic because of nonfusion of the thalamus or many be large and incorporated into the forebrain monoventricle. Hydrocephalus may be present because of aqueductal atresia, especially in cases with midfacial hypoplasia. In alobar holoprosencephaly, there is always a large dorsal cyst. This is a fluid-filled cavity that fills the posterior half of the cranial cavity, so that the holoprosencephalic brain actually only occupies the rostral half of the cranium. At times, the dorsal
cyst herniates through the anterior fontanelle to form a special type of encephalocele found only in holoprosencephaly. This cephalocele has ependymal remnants in its wall to indicate that it arose within the ventricular system and is not an arachnoidal cyst. The origin of the dorsal cyst may be the large suprapineal recess of the third ventricle (298). A single midline, meandering anterior cerebral artery is present, but both middle cerebral arteries are present (566). The deep venous system of the brain also is malformed (567).
cyst herniates through the anterior fontanelle to form a special type of encephalocele found only in holoprosencephaly. This cephalocele has ependymal remnants in its wall to indicate that it arose within the ventricular system and is not an arachnoidal cyst. The origin of the dorsal cyst may be the large suprapineal recess of the third ventricle (298). A single midline, meandering anterior cerebral artery is present, but both middle cerebral arteries are present (566). The deep venous system of the brain also is malformed (567).
In semilobar holoprosencephaly, the intermediate degree of severity, an incomplete hemispheric fissure is seen posteriorly, and the occipital lobes and the occipital horns of the lateral ventricles are well differentiated, indicating that the rostrocaudal gradient of genetic expression in the cerebral cortex is less extensive than in the alobar form. In lobar holoprosencephaly, the mildest form, partial fusion of the hemispheres or sometimes only of the hippocampus, occurs frontally, with complete separation occipitally. The olfactory bulbs may be differentiated but are hypoplastic.
The type of midfacial hypoplasia ranges from mild hypotelorism and hypoplasia of the bridge of the nose to the most extreme form, cyclopia with a single median eye and a proboscis above the median eye instead of a nose (559,568,569). The two most frequent severe variants are related to which wave of mesencephalic neural crest tissue has been affected (293,298,300). If the first streams of mesencephalic neural crest are defective, there is a midline cleft of the lip and palate because of agenesis of the premaxilla and vomer bones, and hypotelorism, but both eyes and both nostrils are differentiated. If the second streams of mesencephalic neural crest are preferentially impaired, cebocephaly results. In this case, the premaxilla is present, but there is a single nare in the midline of the nose because the lateral haves of the nose on each side fuse because of lack of development of the medial halves of each heminare. A single central incisor has been observed in approximately 25% of cases of the autosomal dominant form of holoprosencephaly (544), and this finding also indicates a disruption of the second of the three main waves of mesencephalic neural crest migration. The severity of facial anomalies often reflects the severity of the cerebral malformations (“the face predicts the brain”) but there are many exceptions, and this often-quoted statement by DeMyer et al. (570) was modified by Sarnat and Flores-Sarnat to “the face predicts the neural crest migration” (298) and reversed by Carstens to indicate the importance of neural tube induction of facial mesoderm: “the brain predicts the face” (294).
In cyclopia, the face is marked by a single median orbital fossa and eye with a protruding, noselike appendage above the orbit. Because the medial side of the globe of the eye is formed from the second mesencephalic neural crest migration and the lateral side by the third, the pathogenesis of the median eye is the same principle as the single nare in cebocephaly. Other dysplastic features include polydactyly and cardiac and digestive tract anomalies. Hypotelorism, a median cleft lip, and a nose that lacks its bridge, columella, or septum almost always denote some degree of holoprosencephaly, even if not associated with any of the aforementioned major anomalies (551,568). The diagnosis of hypotelorism should be based on formal measurements of the interpupillary, interorbital, and intercanthal distances (571). The most severe craniofacial malformations are usually associated with the most severe cerebral form, alobar holoprosencephaly, but this is not always the case, and cyclopia and cebocephaly can be seen in semilobar and even in lobar holoprosencephalies.
The term arhinencephaly has been used to describe a variety malformations ranging from isolated absence or hypoplasia of the olfactory bulbs and tracts to the association of this anomaly with the various forms of holoprosencephaly (551,572). Arhinencephaly can be unilateral and associated with hemifacial microsomia and oculoauriculovertebral dysplasia (Goldenhar syndrome) (573). It also can be accompanied by hypogonadotropic hypogonadism (Kallmann syndrome) (574).
Diagnostic studies in holoprosencephaly should include facial radiography to show deformed anterior craniobasal bones, cytogenetics, and MRI for definitive evaluation of the CNS abnormalities. The EEG, visual-evoked potentials, and auditory-evoked potentials are generally abnormal. When the patient has many extracephalic abnormalities, a chromosomal anomaly is likely, whereas in their absence, the karyotype is usually normal. Neuroendocrine studies should be performed to study the function of the hypothalamic–pituitary axis. A simple but often neglected part of the neurologic examination of young infants is a test for olfactory reflexes using a nonirritating substance such as peppermint (not alcohol or ammonia): In most neonates a sucking response is elicited, and in about one-third there is arousal withdrawal; a good and reproducible response indicates that arinencephaly is not present (575).
Septo-Optic-Pituitary Dysplasia
Septo-optic-pituitary dysplasia, first described in 1956 by DeMorsier (576), bears some similarities to holoprosencephaly in being another disorder of midline cleavage and hypoplasia of median diencephalic and telencephalic structures. Some cases are associated with mutations or deletions in the HESX1 gene (577,578), and in others the PAX3 gene has been shown to be defective (579). Septo-optic-pituitary dysplasia includes agenesis of the septum pellucidum, hypoplasia of the optic nerves and chiasm with resultant blindness or severe visual impairment,
hypoplasia of the infundibulum with growth hormone deficiency and short stature, and, in approximately one-third of children, diabetes insipidus (580,581). Although growth hormone deficiency is the most common isolated posterior pituitary insufficiency in septo-optic-pituitary dysplasia, some patients have panhypopituitarism. Endocrine function, therefore, should be evaluated in all children in whom the diagnosis is confirmed by imaging. Psychomotor function can be preserved (582). Optic nerve hypoplasia can occur in the absence of any other developmental anomaly, and as such it is a relatively common congenital anomaly (583). The condition is reviewed by Zeki and Dutton (584).
hypoplasia of the infundibulum with growth hormone deficiency and short stature, and, in approximately one-third of children, diabetes insipidus (580,581). Although growth hormone deficiency is the most common isolated posterior pituitary insufficiency in septo-optic-pituitary dysplasia, some patients have panhypopituitarism. Endocrine function, therefore, should be evaluated in all children in whom the diagnosis is confirmed by imaging. Psychomotor function can be preserved (582). Optic nerve hypoplasia can occur in the absence of any other developmental anomaly, and as such it is a relatively common congenital anomaly (583). The condition is reviewed by Zeki and Dutton (584).
The insult responsible for septo-optic-pituitary dysplasia probably begins at approximately 37 days’ gestation, though underexpression of an organizer gene would begin even earlier. A variety of causes have been recorded, including maternal diabetes, maternal anticonvulsant intake, and cytomegalic inclusion disease (585). Additionally, optic nerve hypoplasia is seen in conjunction with the Klippel-Trenaunay-Weber syndrome, chondrodysplasia punctata, and Kallmann syndrome (hypogonadotropic hypogonadism) (585,586).
Noncleft Median Face Syndrome
Noncleft median face syndrome includes several syndromes familiar to pediatricians, such as Treacher Collins syndrome, Crouzon disease, and Apert syndrome. It also embraces the chromosomal trisomies 18 and 21. The facial deformities are mild but stereotyped, characterized by mongoloid and antimongoloid slants and abnormally spaced eyes (hypertelorism or hypotelorism). In a significant proportion of cases, pathologic examination or neuroimaging studies of the brain reveal maldevelopment of the neocortex with frequent migration anomalies causing defective cortical lamination, and occasional failure of inductive diverticulation.
DISORDERS OF CELLULAR PROLIFERATION
Rarely, severely hypoplastic brains are found that appear to be developmental arrests at early embryonic or fetal stages. Two examples are illustrated in Fig. 5.7. These maturational arrests appear to be a failure of adequate cellular proliferation in the neural tube, and the etiology is probably multiple. One mechanism may be if the ependyma is induced to differentiate too early, so that all of the mitotic cycles at the ventricular wall are not completed (73,587). Whether accelerated apoptosis may be destroying cells as they are formed at an excessive rate is speculative as another mechanism. Congenital viral infections often produce brains that are excessively small and lack the number of neurons and glial cells expected, but this is often the result of microinfarcts due to endothelial cell and vascular involvement. Vaccines in the first trimester of pregnancy also are implicated in some cases, but this is difficult to prove (588).
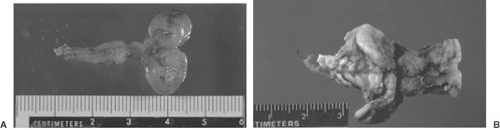 FIGURE 5.7. A. Global cerebral hypoplasia in a 21-week fetus, a maturational arrest probably due to deficient neuronogenesis. The cerebral hemispheres, brainstem and cerebellum are all very small and underdeveloped even for this gestational age. The brain weighed 2.3 g (normal mean for age is 58.0 g). (From Sarnat HB. Cerebral dysgenesis. Embryology and clinical expression. New York: Oxford University Press, 1992. With permission.) B. Dorsal view of brainstem and cerebellum of a full-term neonate, showing an exposed fourth ventricle because the cerebellum is arrested in development while still at the rhombic lip stage or a little beyond. The mother had received a swine influenza vaccine in the first trimester, but whether this was the cause is uncertain. (From Sarnat HB, Rybak G, Kotagal S, et al. Cerebral embryopathy in late first trimester: possible association with swine influenza vaccine. Teratology 1979;20:93–100. With permission.) |
DISORDERS OF CELLULAR MIGRATION (1 TO 7 MONTHS’ GESTATION)
Although disorders of organ induction are known to produce secondary histogenic migratory anomalies, this section is confined to those disorders of cellular migration that are unassociated with defects of embryogenesis. Over the last few years, MRI has permitted diagnosis of these
conditions during the lifetime of the affected child, and it is becoming apparent that their incidence is much greater than had previously been estimated. Sarnat, Barth, and Barkovich and their coworkers have published reviews of these various disorders (73,589,590). Nevertheless, many microdysplasias of the cerebral cortex that are focal migratory disturbances are beneath the limits of resolution of imaging studies and are only seen microscopically. In the past these were poorly understood because autopsy confirmation of the clinical suspicion was rarely available, but it is now with the advent of epilepsy surgery and the availability of surgical brain tissue; these malformations are now being well studied.
conditions during the lifetime of the affected child, and it is becoming apparent that their incidence is much greater than had previously been estimated. Sarnat, Barth, and Barkovich and their coworkers have published reviews of these various disorders (73,589,590). Nevertheless, many microdysplasias of the cerebral cortex that are focal migratory disturbances are beneath the limits of resolution of imaging studies and are only seen microscopically. In the past these were poorly understood because autopsy confirmation of the clinical suspicion was rarely available, but it is now with the advent of epilepsy surgery and the availability of surgical brain tissue; these malformations are now being well studied.
The various conditions associated with migration defects are listed in Table 5.16. They include the phakomatoses; a variety of metabolic, genetic, and chromosomal syndromes; and maternal and environmental causes.
Migratory disorders develop when neuroblasts of the subventricular zone (i.e., the germinal matrix), which forms the wall of the lateral ventricles, fail to reach their intended destination in the cerebral cortex. This results in focal or generalized structural deformities of the cerebral hemispheres. These are discussed in sequence of their ontogenetic chronology.
Schizencephaly
Schizencephaly is characterized by clefts placed symmetrically within the cerebral hemispheres and extending from the cortical surface to the underlying ventricular cavity (Figs. 5.8 and 5.9) (73,589,590,591). The walls of the cleft may be in apposition or separated. The cerebral cortex that surrounds the cleft may be normal or show areas of polymicrogyria. This suggests that in many instances schizencephaly results from pathogenetic processes similar to those that cause polymicrogyria, but are more extensive and involve the entire thickness of the developing cerebral hemispheres (592).
A mutation of the homeobox gene EMX2 has been identified in schizencephaly (98,593). This mutation does not account for all cases, however, and some are not really primary developmental malformations at all, but occur secondary to porencephaly or other encephaloclastic lesions acquired during midfetal life (73).
Schizencephaly should be distinguished from porencephaly caused by a variety of vascular or infectious insults to the brain during late fetal or early infantile life. A porencephalic cyst results from the dissolution of necrotic regions of brain, with cavitation and cyst formation within the parenchyma of the cerebral hemispheres. Porencephalic cysts communicate with the ventricular system or may extend to the cerebral cortical surface but do not destroy the thin pial membrane. Occasionally, they act as a space-occupying lesion, causing symptoms of increased intracranial pressure (594). Most important, they are asymmetric, not aligned with the primary fissures, and unassociated with major cerebral migration defects.
TABLE 5.16 Conditions Associated With Neuroblast Migratory Disorders | ||
|---|---|---|
|
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


