Fig. 5.1
Microtubule biology and cellular function. (a) Tubulin heterodimers are the basic building units of microtubules. Both isoforms can bind to a guanidine nucleotide. Whereas α-tubulin cannot hydrolyze and exchange GTP, β-tubulin can exist in two states: GTP- or GDP-bound. GTP-tubulin is the active state of the heterodimer with regards to polymerization capability (indicated with a star). (b) Tubulin heterodimers build up protofilaments by a juxtapositional head-to-tail arrangement. The β-tubulin is oriented towards the so called plus-end, the α-tubulin towards the minus-end. Protofilaments are arranged into a hollow microtubule and have lateral interactions with each other. Note that protofilaments are not formed per se, but are always part of the microtubule complex. (c) A polymerizing microtubule will add GTP-bound heterodimers, which will hydrolyze their nucleotide. This results in a GTP-tubulin cap at the plus-end of the microtubule. Due to low concentrations (in vitro) or the activity of certain accessory proteins the microtubule can change into a depolymerizing state; this transition is called catastrophe. (d) Depolymerizing microtubules are characterized by curved protofilaments (a structural trait of GDP-bound heterodimers) at the plus-tip end that leave the microtubule complex. The reversal of this state into a polymerizing microtubule is called rescue. (e) Rigid microtubules are used as transport highways for different cargo, such as macromolecular protein complexes or vesicles. Specialized motor proteins can attach to the microtubule lattice and show processive movement towards the plus- or the minus-end; these motors will bind to cargo and transport it along the microtubule. (f) Dynamic microtubules are needed for chromosome alignment in the metaphase plate (upper panel) and for sister chromatid separation during anaphase (lower panel). MTOC Microtubule Organizing Center, centrosome. Modelled on Akhmanova and Steinmetz (2008, 2010), Conde and Caceres (2009), Dogterom et al. (2005), Kuijpers and Hoogenraad (2011) and Schliwa and Woehlke (2003)
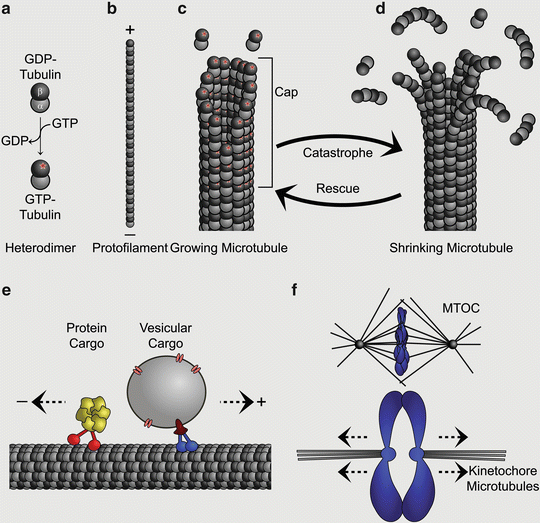
Fig. 5.2
Examples for microtubule functions in neurodevelopment. (a) Schematic of the developing cortex. Radial glial progenitors span the entirety of the cortex from the ventricular to the pial surface (shown in orange). They undergo mitosis to generate other types of progenitors (not shown) and neurons (shown in yellow). Postmitotic neurons migrate along their radial glial mother cell to their final destination in the cortical plate where they will differentiate and extend their axons. (b) Detailed view of the interkinetic nuclear migration of a radial glial cell. The cell nuclei migrate basally (upwards) during G1 phase, undergo S phase and migrate apically (downwards) during G2. Finally, cells will undergo mitosis at the ventricular surface. Microtubules are required for interkinetic nuclear migration and spindle formation (shown in green) which mediates sister chromatid separation in M phase. (c) Neuronal migration requires nuclear translocation. Nuclei are surrounded by a microtubule cage (nuclear cage; shown in green) that connects with the actomyosin network (shown in red) via the centrosome. (d) Differentiating neurons extend their neurites to form connections. Axonal projections have to cover large distances within the brain and the correct pathfinding requires the establishment of a growth cone. This specialized structure consists of microtubules (shown in green) that provide a rigid platform which interacts with the actin cytoskeleton (shown in red), facilitating the extension of the lamellipodia
2 Tubulin Diversity and Genetics
Shortly after this conference, it became apparent that the α- and β- isoforms were not single entities, but rather existed in multiple flavours. N-terminal sequencing and biochemical separation experiments led Hayashi and colleagues to conclude that both types of tubulins include multiple isotypes consisting of slightly different amino acids (Bryan et al. 1978). The advent of molecular cloning further expanded the tubulin family. cDNA libraries constructed by Cowan and colleagues from chicken brain mRNA resulted in the identification of four α- and four β-tubulin genes; a list which has grown over the years (Krauhs et al. 1981; Lopata et al. 1983; Cleveland et al. 1978, 1980; Cowan et al. 1981; Wilde et al. 1982a, b; Cowan and Dudley 1983; Hall et al. 1983; Little et al. 1981; Ponstingl et al. 1981). With the completion of the human and mouse genome sequences, we now know that there are seven α- and eight β-tubulins in mice; and eight α- and nine β-tubulins in humans (Table 5.1).
Table 5.1
List of all human and Murine Tubulin-Isotypes
Symbol | Name | NCBI Gene ID | |||
|---|---|---|---|---|---|
Mouse | Human | Mouse/Human | Mouse | Human | |
α-Tubulins | Tuba1a | TUBA1A | Tubulin, α 1A | 22142 | 7846 |
Tuba1b | TUBA1B | Tubulin, α 1B | 7846 | 10376 | |
Tuba1c | TUBA1C | Tubulin, α 1C | 22146 | 84790 | |
Tuba3a | – | Tubulin, α 3A | 22144 | – | |
Tuba3b | – | Tubulin, α 3B | 22147 | – | |
– | TUBA3C | Tubulin, α 3C | – | 7278 | |
– | TUBA3D | Tubulin, α 3D | – | 113457 | |
– | TUBA3E | Tubulin, α 3E | – | 112714 | |
Tuba4a | TUBA4A | Tubulin, α 4A | 22145 | 7277 | |
Tuba8 | TUBA8 | Tubulin, α 8 | 53857 | 51807 | |
β-Tubulins | Tubb1 | TUBB1 | Tubulin, β 1 Class VI | 545486 | 81027 |
Tubb2a | TUBB2A | Tubulin, β 2A Class IIA | 22151 | 7280 | |
Tubb2b | TUBB2B | Tubulin, β 2B Class IIB | 73710 | 347733 | |
Tubb3 | TUBB3 | Tubulin, β 3 Class III | 22152 | 10381 | |
Tubb4a | TUBB4A | Tubulin, β 1 Class VI | 22153 | 10382 | |
Tubb4b | TUBB4B | Tubulin, β 4B Class IVB | 227613 | 10383 | |
Tubb5 | TUBB5 | Tubulin, β 5 Class I | 22154 | 203068 | |
Tubb6 | TUBB6 | Tubulin, β 6 Class V | 67951 | 84617 | |
– | TUBB8 | Tubulin, β 8 Class VIII | – | 347688 | |
With the exception of the carboxy terminus, the α- and β-tubulin isoforms exhibit a high degree of sequence homology; however, their expression pattern varies (Lewis et al. 1985; Villasante et al. 1986; Wang et al. 1986; Burgoyne et al. 1988). For instance, in humans the β-tubulin isoform TUBB1 is specifically found in platelets and megakaryocytes, whereas TUBB3 is expressed in post-mitotic neurons (Wang et al. 1986; Schulze et al. 2004; Liu et al. 2007). Similarly, in Arabidopsis thaliana, the ArathTub9 isoform accumulates specifically in male reproductive tissue, the pollen, whereas ArathTub1 is preferentially found in roots and leaves (Oakley et al. 2007; Snustad et al. 1992; Cheng et al. 2001).
3 The Multi-tubulin Hypothesis
The existence of this extended gene family with distinct expression patterns led investigators to speculate that the different tubulin isoforms possess unique functional properties, accounting for the extraordinary diverse role microtubules play in eukaryotic cells (Fulton and Simpson 1976). This concept, which is referred to as the multi-tubulin hypothesis, was advanced by Raff and co-workers who employed the genetic tools available in Drosophila to replace the testes specific tubulin β-2 with the developmentally expressed β-3 tubulin (Kemphues et al. 1979). The β-3 isoform could support the assembly of a cytoskeletal array, but the substitution nevertheless resulted in defects of axoneme structure, meiosis, and nuclear shaping (Hoyle and Raff 1990; Raff et al. 1997). The Raff group was further able to show that the architecture of microtubules was influenced by the isoform composition. Transgenic expression of the moth β-tubulin (Hvβt) alongside the β-2 isoform resulted in a Drosophila germline that was dominated by microtubules with 16 protofilaments, not the usual 13 (Hoyle and Raff 1990; Raff et al. 1997).
Parallel to these reports a mutagenesis screen performed in the group of Martin Chalfie identified a specific β-tubulin isoform (MEC7) that caused loss of touch receptivity in the nematode worm C. elegans (Savage et al. 1989, 1994). Similar to Raff and colleagues, they observed that the isoform composition of microtubules could affect the microtubule superstructure. MEC-7 mutants showed a shift from microtubules with 15 protofilaments to microtubules with just 11 protofilaments. This result was mirrored by another C. elegans strain harboring mutations in the α-tubulin MEC-12, which is also highly expressed in touch-sensitive neurons and is believed to co-assemble with MEC-7. Mutations in this tubulin again resulted in the loss of microtubules with 15 protofilaments (Fukushige et al. 1999).
These findings suggested that the protofilament number is a fixed inherent property of microtubules which is dependent on the tubulin composition. This is supported by the finding that the predominant configuration of mammalian microtubules in cells is 13 protofilaments (McIntosh et al. 2009; Tilney et al. 1973). However, in vitro experiments have shown that vertebrate tubulin-heterodimers by themselves assemble into microtubules ranging from 8 to 17 protofilaments (Chretien et al. 1992; Chretien and Wade 1991). Therefore, preference for a specific number of protofilaments for one isoform can only be determined by interaction with factors present in vivo.
So, what factors determine the protofilament number? Brouhard and colleagues have demonstrated that the microtubule associated protein DCX (Doublecortin) stabilizes the 13 protofilament configuration in vitro and they argue that this might be one of the main mechanisms that ensure correct protofilament numbers in neurons (Bechstedt and Brouhard 2012). Likewise, the nucleation by a γ-tubulin ring complex contributes to a consistent width of cellular microtubules (Moritz et al. 2000). Posttranslational modifications may also play a role. Goodman and colleagues have shown that acetylation of the α-tubulin MEC-12 stabilizes the 15 protofilament configuration found in C. elegans touch receptive cells (Cueva et al. 2012). The deletion of the responsible acetylase, ATAT-2, results in highly variable protofilaments numbers. This observation has led Goodman and colleagues to the proposition that acetylation promotes the formation of salt bridges that mediate lateral interactions between protofilaments (Cueva et al. 2012). In their model an interaction between glutamate at position 55 and lysine 40 exists within the α-tubulin (αE55-αK40). This salt bridge is disrupted by acetylation of the K40 residue, favoring an interaction between adjacent heterodimers (αE55-α′H283), the angle of which is consistent with 15 protofilament microtubules (Cueva et al. 2012).
4 Posttranslational Modifications
Acetylation is but one of a myriad of different posttranslational modifications associated with the tubulins. Others include detyrosination, polyglutamylation, polyglycylation, palmitolyation and phosphorylation (Janke and Bulinski 2011; Westermann and Weber 2003). These modifications affect tubulin dynamics and stability, the interaction with motor proteins and also non-motor microtubule associated proteins. The amino acid sequence of individual tubulin isoforms influences their respective posttranslational modifications. For instance, in mice and humans TUBA8 lacks a lysine at the critical residue 40, and consequently cannot be acetylated. This contrasts with the remaining members of the α-tubulin family, all of which have this residue, and therefore can be subject to this modification (Fukushige et al. 1999; Stanchi et al. 2000). Likewise, some tubulin isoforms, such as the testis specific cα2 in chicken, lack a carboxy-terminal tyrosine residue and are therefore not subject to detyrosination (Pratt et al. 1987).
5 Tubulin proteins in Neurodevelopmental Disease – The Makers
Since their discovery in the 1950s it is has been clear that microtubule function is essential for the formation and function of the nervous system in a broad range of animal species, whether it be a nematode, a fruit fly, a frog or a rodent (Goldstein and Yang 2000; Gerson et al. 1976; Gray 1975, 1976; Ward et al. 1975; Poulain and Sobel 2010). It is no surprise that the same holds true for the development of the human brain. Microtubules facilitate neurogenic division, they drive neuronal migration, and they are required for neuronal differentiation and circuit formation (Ayala et al. 2007; Kuijpers and Hoogenraad 2011) (Fig. 5.2). Here we discuss the role of the different tubulins in these processes with a focus on human diseases caused by mutations in these genes (Fig. 5.3).
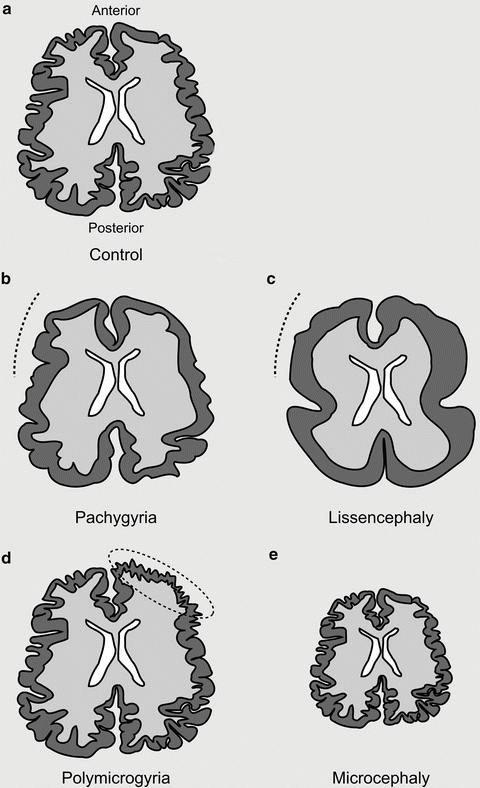
Fig. 5.3
Spectrum of tubulinopathies. (a) Depiction of an axial section of a control brain with regular distribution and number of sulci and gyri. (b) Pachygyric (meaning thick gyri) patients show a reduction in the number and increase in the size of their gyri (indicated with a dotted line). (c) Lissencephalic (meaning smooth brain) patients show a complete absence of sulci and gyri. Both pachygyria and lissencephaly can be present as a gradient from anterior to posterior. (d) Polymicrogyric (meaning many small gyri) patients show an increase in the number of gyri with a decreased size. This is often focally localized and asymmetric. (e) Microcephalic (meaning small head) patients show a reduction in overall brain size (−2SD from the mean). Microcephaly vera (or primary microcephaly) occurs in the absence of other cortical malformations
6 TUBA1A – The First
The tubulin gene family was first implicated in neurodevelopmental disease following the cloning of an N-ethyl-N-nitrosourea (ENU) induced Tuba1a mutation in the Jenna mouse mutant (Keays et al. 2007). It was identified in a screen for hyperactive behavior, but also showed defects in working memory and presented with an exaggerated acoustic startle response (Edwards et al. 2011; Keays et al. 2007, 2010). Histological examination revealed wave-like perturbations of the adult cortex, a fractured pyramidal layer of the hippocampus and structural abnormalities in the superior colliculus; defects which were attributed to impaired neuronal migration. These phenotypes were reminiscent of mouse models of lissencephaly (Lis1, Dcx, and the Reeler mouse), a disease which is characterized by a cortex with a smooth surface (Gleeson and Walsh 2000; Guerrini and Parrini 2010) (see also Chap. 1). Speculating that mutations in TUBA1A might cause neurodevelopmental disease in humans, a genetic screen identified two de novo mutations in this gene (R264C and R402H) in patients with lissencephaly (Keays et al. 2007).
The introduction of a TUBA1A genetic test into clinical practice has resulted in the identification of a host of disease causing mutations in this gene (Poirier et al. 2007, 2012; Fallet-Bianco et al. 2008; Bahi-Buisson et al. 2008; Morris-Rosendahl et al. 2008; Kumar et al. 2010; Lecourtois et al. 2010; Jansen et al. 2011; Mokanszki et al. 2012; Sohal et al. 2012; Hikita et al. 2013). Most patients identified have de novo mutations and present with a spectrum of phenotypes that extends from an absence (agyria), to a reduction (pachygyria) or even an increased number of gyri (polymicrogyria) (Poirier et al. 2007, 2012; Kumar et al. 2010). These cortical phenotypes are frequently accompanied by hypoplasia or agenesis of the corpus callosum, hypoplasia of the brain stem, dysgenesis of the basal ganglia, ventricular dilation, and hypoplasia of the cerebellum (Sohal et al. 2012; Kumar et al. 2010). In addition, almost all patients with TUBA1A mutations present with a reduction in brain size (−1 S.D. to −7 S.D. from mean), most classifying as microcephalic (less than −2 S.D. below mean; more than 90 %) (Poirier et al. 2007, 2012; Sohal et al. 2012; Kumar et al. 2010).
7 Molecular and Cellular Mechanisms of TUBA1A Mutations
What is the underlying molecular defect that results in the disease state in patients with TUBA1A mutations? TUBA1A, like all tubulins protein, has three major domains; an N-terminal domain (1–229), an intermediate domain (230–371), and a C-terminal domain (372–450). The N-terminal domain harbors a GTP binding pocket that, in the case of α-tubulins, is non-exchangeable and is thought to act as a structural co-factor (Nogales et al. 1997; Spiegelman et al. 1977). In the case of the Jenna mouse it was shown that the S140G mutation caused impairment in GTP binding, and, consequently, a dramatic reduction in heterodimer formation. The mutant heterodimers, however, were able to incorporate into the microtubule cytoskeleton, suggesting that the mutation acted by haploinsufficiency. Similarly, the human mutations V303G, L397P, and R402C all result in a reduction in heterodimer levels, which have been attributed to molecular defects in the tubulin folding pathway (Tian et al. 2008, 2010). It is apparent, however, that some disease causing tubulin mutations have no effect on the efficiency of chaperon mediated tubulin folding whatsoever. For instance, in vitro analysis of the P263T, L286F, R402H, and S419L mutations has shown that they do not cause impaired heterodimer folding. In the case of the P263T mutation the incorporation of mutant heterodimers into the microtubule lattice has a deleterious effect of microtubule dynamics and growth, lending itself to the conclusion that some tubulin mutations act by a dominant negative mechanism (Tian et al. 2010). Mutations that fall within this class may influence the binding of microtubule associated proteins such as DCX or the kinesins (Amos and Schlieper 2005). Tubulins might also interact with unknown microtubule associated proteins that are vital for the formation of the developing brain.
What are the underlying cellular mechanisms that give rise to TUBA1A-related disease? In addressing this question it is important to appreciate that TUBA1A is highly expressed in post-mitotic neurons, but not glia, in the human and mouse brain (Gloster et al. 1999; Bamji and Miller 1996). Murine expression studies have shown that TUBA1A is largely absent from the proliferative ventricular zone (VZ), and its expression peaks at embryonic day (E) 16.5 (Braun et al. 2010). The migration of neurons requires the extension of the leading process, the translocation of the nucleus and the retraction of the trailing process (Trivedi and Solecki 2011) (see also Chaps. 1, 2, 4 and 7). All of these processes are heavily reliant on a dynamic microtubule network, and could potentially be impaired by mutations in TUBA1A. Similarly, neurite outgrowth requires the stable support and dynamic force generated by microtubules (Dent and Gertler 2003). Defects in this process can cause inadequate crossing of the midline, resulting in an abnormal corpus callosum and neurological defects (Engle 2010). Disorders of axon guidance or migration, however, fail to account for the reduction in brain size that is observed in almost all patients with mutations in TUBA1A. This is particularly curious, given its post-mitotic expression. One explanation that might account for this phenotype is an increase in neuronal apoptosis, which has been observed in the adult superior colliculus in the Jenna mouse (Edwards et al. 2011). This explanation is consistent with the observation that TUBA1A associated microcephaly can increase in severity postnatally (Cushion et al. 2013).
Why do mutations in TUBA1A cause a spectrum of distinct neurological disorders? Initially this gene was strongly associated with lissencephaly/pachygyria, but it is now clear that de novo mutations can also cause polymicrogyria. For instance, a mutation in valine 235 (V235L) results in bilateral and asymmetric polymicrogyria, whereas mutations in arginine 402 (R402C, R402H) cause classic lissencephaly (Mokanszki et al. 2012; Kumar et al. 2010; Poirier et al. 2007). An analysis of the position of polymicrogyria and lissencephaly causing mutations reveals no obvious pattern (Fig. 5.4). It is conceivable that different diseases are a consequence of defects in different cellular processes associated with microtubule based neuronal migration. However, this would not account for the interesting case of the R390C mutation. This very same mutation has been reported to cause polymicrogyria in a 1-year-old boy and mild gyral simplification and total agenesis of the corpus callosum in another child (Poirier et al. 2012; Kumar et al. 2010). How does the same mutation cause two distinct migration phenotypes? One possibility could be the exposure to different environmental conditions in utero; or additional genetic factors that contribute to one or the other phenotype.

Fig. 5.4
Mutations associated with TUBA1A
8 TUBB2B – Expanding the Spectrum
Given that mutations in TUBA1A cause neurodevelopmental disease, it was reasonable to speculate that mutations in the β-tubulins might also be pathogenic. Following a genetic screen of TUBB2A, TUBB2B, and TUBB2C, the Chelly group reported the identification of five cases of asymmetrical polymicrogyria (four patients, one aborted fetus) caused by mutations in TUBB2B (Jaglin et al. 2009). Besides asymmetrical polymicrogyria, each patient presented with additional features, such as microcephaly, dysmorphic basal ganglia, cerebellar dys- or hypoplasia, abnormal corpus callosum and brain stem hypoplasia. Similar to TUBA1A, the spectrum of TUBB2B related diseases has expanded rapidly. Engle and colleagues recently reported the occurrence of a mutation (E421K) that causes congenital fibrosis of the extraocular muscles (CFEOM), a specific defect of axon guidance, accompanied by polymicrogyria (Cederquist et al. 2012); axon guidance defects accompanied by polymicrogyria and schizenecephaly have also been reported for a G140A mutation (Romaniello et al. 2012). Pilz and colleagues have described a lissencephalic patient with a TUBB2B mutation (L207P), and Guerrini and colleagues have reported an individual with pachygyria and microcephaly with an N256S mutation (Cushion et al. 2013; Guerrini et al. 2012) (Fig. 5.5). To date biochemical analysis has been conducted on five TUBB2B mutations (F265L, I210T, L228P, S172P and T312M) and, similar to TUBA1A mutations, they influence tubulin heterodimer folding and their incorporation into microtubules in different ways. For instance, the S172P mutation results in arrested tubulin heterodimer folding, whereas the I210T is indistinguishable from the wild-type in biochemical and cellular assays.
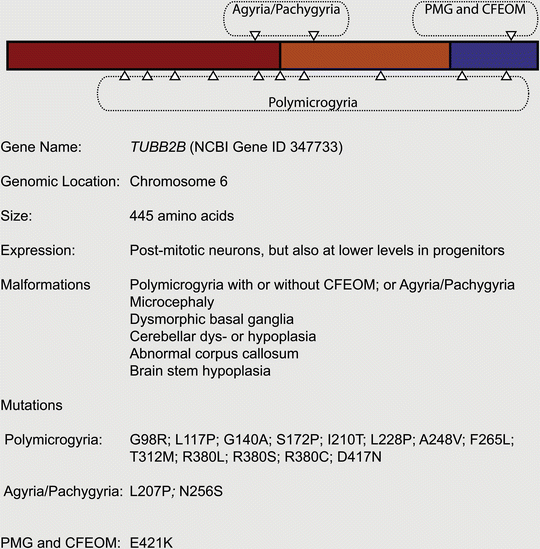
Fig. 5.5
Mutations associated with TUBB2B
As might be expected TUBB2B is highly expressed in post-mitotic neurons at key developmental time-points, but is also found in progenitor cells at lower levels (Jaglin et al. 2009). In vivo knockdown experiments in the rat have shown that Tubb2b is required for radial migration. These data have led to the hypothesis that TUBB2B-related cortical malformations are due to a combination of impairment in neuronal migration and radial glial dysfunction (Jaglin et al. 2009).
9 TUBB3 – The Janus Tubulin
The list of tubulinopathy causing genes expanded in 2010 with the addition of TUBB3 by two independent studies. Engle and colleagues showed that six different heterozygous mutations in this gene caused congenital fibrosis of the extraocular muscles type 3 (CFEOM3), either in isolation or as a component of a syndrome (Tischfield et al. 2010). Interestingly, and in marked contrast to the previously described TUBB2B and TUBA1A mutations, no neuronal migration deficits or microcephaly could be observed in these patients. The pathogenicity of one mutation (R262C) was explored further by the creation of a transgenic mouse line that replicated various aspects of the human disease. The R262C mutation increased microtubule stability and impaired their interaction with the motor protein Kif21a. Ultimately, this resulted in defects of axon guidance and cranial nerve extension, but not cortical architecture (Tischfield et al. 2010). In the same year, the Chelly group reported six different TUBB3 mutations (five heterozygous, one homozygous) in nine patients with malformations of cortical development associated with neuronal migration defects (Poirier et al. 2010). All patients suffered from polymicrogyria or gyral disorganization with microcephaly and cerebellar dys- or hypoplasia. With the exception of one individual, these patients presented with brainstem hypoplasia, an abnormal corpus callosum and dysmorphic basal ganglia, but not the CFEOM3 phenotypes described by Engle and colleagues (Fig. 5.6). None of the mutations identified by the Chelly group were the same as those that cause CFEOM3; curiously, however, both studies described different mutations in the same residue, A302. Its substitution with a threonine caused CFEOM3, whereas the A302V mutation was pathogenic in a patient with gyral disorganization (Tischfield et al. 2010; Poirier et al. 2010). These data imply that the mutations probably act by a dominant mechanism and not by haploinsufficiency. This idea is supported by the observation that mutations that cause cortical malformations, in contrast to the ones causing CFEOM3, reduce microtubule stability (Tischfield et al. 2010; Poirier et al. 2010). Finally, it should be noted that the phenotypes associated with TUBB3 have recently been expanded to include peripheral neuropathy, defective olfactory function, photophobia, cyclic vomiting and hypogonadotropic hypogonadism with analogies to Kallmann syndrome (Chew et al. 2013).

Fig. 5.6
Mutations associated with TUBB3
10 TUBB5 – The Mitotic Tubulin
We have added another tubulin isoform to the list of disease-causing genes: TUBB5 (Breuss et al. 2012). In contrast to the other tubulinopathies, the primary defect associated with TUBB5 is microcephaly. We reported three unrelated individuals with de novo TUBB5 mutations (M299V, V353I, E401K) with only one patient presenting with a notable migration phenotype (M299V) (Fig. 5.7). Similar to other tubulin isotype-dependent disorders, affected individuals presented with dysmorphic basal ganglia and corpus callosum abnormalities. Employing a transgenic mouse line that expresses GFP under the endogenous Tubb5 promoter, we have shown that TUBB5 is expressed in radial glial progenitors, intermediate progenitors, and post-mitotic neurons. Depletion of TUBB5 in utero by shRNA knockdown perturbed the cell cycle of progenitors and resulted in neuronal migration defects. Similarly, we have found that overexpression of two of the three TUBB5 mutations (E401K and V353I) increased the percentage of progenitors in M-phase and altered neuronal positioning. Intriguingly, these two mutations affected the tubulin folding pathway in different ways. The behavior of the V353I mutation was indistinguishable from wild-type tubulin, whereas the E401K mutation disrupted the chaperone-mediated folding with a consequent dearth of α/ß heterodimers that failed to incorporate into the cytoskeletal network. This result highlights that tubulin mutations that operate by different mechanisms can still result in similar phenotypes.

Fig. 5.7
Mutations associated with TUBB5
11 TUBB4A – Postnatal and Motor-Related
There is emerging evidence that the tubulinopathies are not limited to developmental phenotypes. In 2013 two independent groups reported the cloning of an R2G mutation in TUBB4A in a multigeneration Australian family that suffered from Whispering Dysphonia. Affected individuals in this family presented with a characteristic “hoppy horse” gait, laryngeal dysphonia, and a thin face (Hersheson et al. 2012; Lohmann et al. 2012). Klein and colleagues additionally described an A271T mutation in an unrelated familial case of segmental dystonia with spasmodic dysphonia (Lohmann et al. 2012). Complementing this finding, Vanderver and colleagues have reported that D249N mutations in TUBB4A cause a rare form of hereditary leukoencephalopathy, characterised by hypomyelination with atrophy of the basal ganglia and the cerebellum (H-ABC) (Simons et al. 2013) (Fig. 5.8). Most of the affected individuals, which originated from seven independent families, presented in infancy with motor dysfunction, but with normal cognitive and language development. While the underlying cellular and molecular mechanisms responsible for these phenotypes remain to be defined, it is known that the Asp249 residue forms a salt bridge with Arg2. This is important for the correct positioning of the T7 loop that interacts with the α-tubulin bound GTP. It is therefore a tenable hypothesis that disruption of this bridge impairs heterodimer stability or microtubule dynamics. Given the postnatal motor-deficits in those individuals with TUBB4A mutations, it is an unsurprising fact that this gene is expressed at low levels in the developing CNS, but is highly transcribed in adult cerebellum, brainstem and striatum (Breuss et al. 2012; Leandro-Garcia et al. 2010). It remains to be determined which cell types in the adult brain express this gene.

 The Impact of JNK on Neuronal Migration
The Dynamics of Neuronal Migration
Spinal Motor Neuron Migration and the Significance of Topographic Organization in the Nervous System
Mark/Par-1 Marking the Polarity of Migrating Neurons
The PAR Polarity Complex and Cerebellar Granule Neuron Migration
Extracellular Signals Controlling Neuroblast Migration in the Postnatal Brain
The Impact of JNK on Neuronal Migration
The Dynamics of Neuronal Migration
Spinal Motor Neuron Migration and the Significance of Topographic Organization in the Nervous System
Mark/Par-1 Marking the Polarity of Migrating Neurons
The PAR Polarity Complex and Cerebellar Granule Neuron Migration
Extracellular Signals Controlling Neuroblast Migration in the Postnatal Brain




Related posts:
The Impact of JNK on Neuronal Migration
The Dynamics of Neuronal Migration
Spinal Motor Neuron Migration and the Significance of Topographic Organization in the Nervous System
Mark/Par-1 Marking the Polarity of Migrating Neurons
The PAR Polarity Complex and Cerebellar Granule Neuron Migration
Extracellular Signals Controlling Neuroblast Migration in the Postnatal Brain
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
