Fig. 1.1
Developmental sequence of inside-out cortical layer assembly. During cortical layer formation, earlier born neurons occupy lower layers and later born neurons progressively occupy upper layers. In the developing mouse cortex, NESCs initially divide mostly symmetrically in the VZ to expand the stem cell pool. The first asymmetric divisions produce cells that occupy the PP. Subsequently, the earliest neurons (purple, future layer VI) forming the CP, split the PP into the SP and MZ, and settle at appropriate positions. The next wave of neurons (dark blue, future layer V), migrate from the VZ/SVZ through the IZ and settle in the CP above the previously generated neurons (purple, layer VI). This developmental sequence is repeated for the red (layer IV), cyan (layer III) and green (layer II) neurons until all cortical layers have been established. During the first three postnatal weeks, a phase of consolidation takes place where cortical neurons finish axon and dendrite genesis, form synaptic connections and assemble into microcircuits. Abbreviations: VZ ventricular zone, SVZ subventricular zone, PP preplate, SP subplate, MZ marginal zone, IZ intermediate zone, CP cortical plate, WM white matter, I–VI cortical layers 1–6, NESCs neuroepithelial stem cells, RGPCs radial glia progenitor cells
2 The Sequential Steps of Cortical Projection Neuron Migration
Nascent cortical projection neurons migrate in a step-wise fashion from their birth place in the VZ/SVZ through the intermediate (IZ) zone in order to reach the cortical plate (CP) and settle at appropriate positions to build up the six cortical layers (Ayala et al. 2007; Marin et al. 2010). While in the rodent the migration paths are still in the range of a few hundred microns, in the developing human cerebral cortex, radially migrating neurons travel approximately 2 cm (Bystron et al. 2008; Meyer 2007; Molnar et al. 2006). Migration routes across the developing cortical wall can be complex (Kriegstein and Noctor 2004; Nadarajah et al. 2003; Tabata and Nakajima 2003) and it has become increasingly clear that radial migration of cortical projection neurons occurs in a tightly regulated manner. Time-lapse and videomicroscopy approaches (Noctor 2011; Tabata and Nakajima 2008; Tsai and Vallee 2011) with the goal to trace the migration paths of individual cortical projection neurons have revealed that (1) radially migrating neurons proceed though several distinct migratory phases; (2) change their morphology along the way and (3) adjust their mode of migration while transiting through the different zones along the radial migratory path (Nadarajah et al. 2001; Noctor et al. 2004; Sekine et al. 2011; Tabata and Nakajima 2003; Tsai et al. 2005).
At early stages of corticogenesis, the migration distances for newly-born neurons are still short but as development continuously progresses, nascent neurons migrate along progressively longer distances. Therefore the patterns of neuronal migration are somewhat different during early versus late corticogenesis (Nadarajah and Parnavelas 2002). The earliest postmitotic neurons delaminate and migrate away from the ventricular surface primarily by somal translocation – by pulling up the soma in the vertical direction with a process stably attached to the pial surface – and form the transient preplate (PP) structure (Allendoerfer and Shatz 1994; Nadarajah et al. 2001; Price et al. 1997; Super et al. 1998). The next wave of postmitotic neurons moves toward the pial surface and splits the PP into the marginal zone (MZ) and the subplate (SP), thus establishing the first neuronal alignment as cortical plate (CP). The CP expands in the vertical direction as consecutive waves of neurons contribute successively to all the layers VI-II in an inside out fashion (Fig. 1.1) (Angevine and Sidman 1961; McConnell 1995; Polleux et al. 1997; Rakic 1974).
During the stages of progressive CP aggregation, nascent cortical projection neurons generated in the proliferative VZ undergo a series of sequential migration steps until they reach their final destination in the CP (Fig. 1.2). First, neurally committed cells delaminate/detach from the neuroepithelium at the ventricular surface (Itoh et al. 2013) and move radially away to the SVZ. Within the SVZ neurons ‘sojourn’ for about 24 h or longer and most adopt a multipolar morphology, extending and retracting processes in all directions (Noctor et al. 2004; Tabata and Nakajima 2003). During this phase, multipolar neurons tend to migrate tangentially in an apparent random fashion (Jossin and Cooper 2011; Noctor et al. 2004) while critical signaling cues induce polarization to predetermine the future axon of the neuron (Barnes and Polleux 2009) (see also Chap. 6). A substantial fraction but not all neurons move retrogradely back towards the VZ before they transform their shape, take on their bipolar morphology and enter the next phase in their migration journey (Noctor et al. 2004). Subsequently, migrating neurons with bipolar morphology locomote along radial glial fibers (Nadarajah et al. 2001; Noctor et al. 2004; Rakic 1972). During this stage of cortical projection neuron migration, the radial movement proceeds in a saltatory fashion by leaps rather than gradual transitions. Cortical projection neurons in the locomotion mode repeat several basic events which underlie their progressive advancement: (1) rapid extension and retraction of the leading neurite which protrudes several dozens of microns from the soma; (2) formation of one or more unsteady swellings/dilatations of the plasma membrane in the leading process; (3) advancement of the centrosome towards and/or into the swelling; (4) forward displacement of the nucleus and soma, a process also known as nucleokinesis; and (5) retraction of the trailing cytoplasmic region and basal process which usually occurs concurrently with nuclear translocation in step 4 (Schaar and McConnell 2005; Tsai and Gleeson 2005; Vallee et al. 2009) (see also Chaps. 2, 4 and 7). Neurons in locomotion mode travel through the IZ until they reach their appropriate target area, the CP. Below the CP, radially migrating neurons have to first pass the SP and then need to invade the earlier generated neurons that occupy the deepest cortical layers. Since the environment of the CP differs significantly from the one in the IZ, especially regarding the density and arrangement of neurons, it is conceivable that the transition from the IZ into the CP must be tightly regulated. The regulatory mechanisms that control the passage of locomoting neurons from the IZ into the CP are however mostly unknown but may involve instructive or simply permissive guidance cues that could act as a gatekeeper for cortical projection neurons to enter their prospective CP target zone. In a next step, neurons then move within the CP further towards the outermost MZ. Once the leading edge of the apical neurite of the locomoting neuron reaches the MZ, the soma with the nucleus rapidly moves up to the top of the CP while the tip of the apical process remains stably attached to the MZ (Nadarajah et al. 2001; Sekine et al. 2011). This last step – terminal somal translocation – is critical for migrating cortical projection neurons to move past all their predecessors in order to ensure appropriate establishment of the inside out lamination within the CP. Recent observations indicate that the uppermost part of the CP comprises of distinct features than the lower CP and was termed primitive cortical zone (PCZ) (Sekine et al. 2011). Interestingly, time-lapse analyses revealed that locomoting neurons seem to transiently pause just below the PCZ and switch at this stage into the terminal translocation mode (Sekine et al. 2011). Once cortical projection neurons have reached their final destination they complete their global differentiation program, finish axon and dendrite genesis, commence synaptogenesis and assemble into microcircuits.
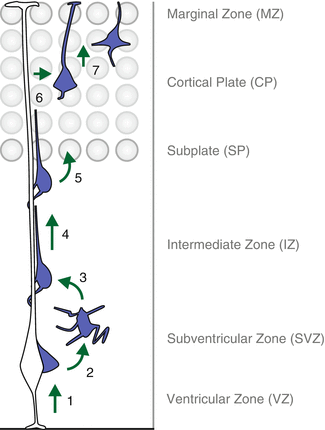
Fig. 1.2
The sequential steps of cortical projection neuron migration. Recent live-imaging experiments (see main text for details) have traced the path of migrating cortical projection neurons and revealed multiple discrete steps and phases along the entire migration journey: (1) nascent neurons delaminate from the ventricular surface in the VZ and move to the SVZ where they adopt a multipolar configuration (2). Upon multi-to-bipolar transition (3), neurons engage in locomotion along the radial glia fiber (4) until they reach the SP, enter the CP target area (5) and continue to migrate towards the MZ. Once the cortical projection neurons have reached the uppermost area of the CP, they detach from the radial glia fiber (6) and execute terminal somal translocation (7) to conclude the radial migration and settle in their appropriate position
3 Role of the LIS1/NDEL1-Complex in the Regulation of Discrete Steps During Cortical Neuron Migration
The importance for appropriate execution of the migration program in cortical projection neurons during brain development is highlighted in patients that suffer from neuronal migration disorders, such as isolated lissencephaly sequence (ILS) or Miller-Diecker Syndrome (MDS) (Dobyns and Truwit 1995; Gleeson and Walsh 2000; Guerrini and Parrini 2010; Pilz and Quarrell 1996; Ross and Walsh 2001). Lissencephaly is characterized by smooth brain surface, abnormal brain morphology and function; and lissencephaly patients suffer from mental retardation and epilepsy. About 40 % of ILS and virtually 100 % of MDS cases occur due to the loss of one copy of the gene called Lissencephaly-1 (LIS1, also known as PAFAH1B1) on human chromosome 17 (Cardoso et al. 2003; Lo Nigro et al. 1997; Pilz et al. 1998; Reiner et al. 1993). Lissencephaly is thus autosomal dominant but the concrete underlying basis of the clinical symptoms, and the mechanisms by which a reduction of LIS1 protein results in human lissencephaly are not well understood. It is however evident that reduced levels of LIS1 negatively affect radial neuron migration (Wynshaw-Boris 2007). Interestingly, it has been documented that increased expression of LIS1 in the developing brain also leads to brain abnormalities in mice and human (Bi et al. 2009). Below, I will first describe the LIS1 protein-interactome and then elaborate on its function in the sequential steps of cortical projection neuron migration in vivo.
3.1 The LIS1/NDEL1-Complex and Its Interacting Partners
The LIS1 protein is evolutionarily conserved from yeast to man and was first identified as one of the nuclear distribution mutants (NudF – for nuclear distribution F) in the filamentous fungus Aspergillus nidulans (Morris 2000; Xiang et al. 1995) amongst other mutants displaying also defective nuclear distribution including NudA, NudC and NudE (Efimov and Morris 2000; Xiang et al. 1999). Orthologues of Lis1 have been identified and cloned in many organisms including Drosophila (Liu et al. 2000) and mice (Hirotsune et al. 1997) whereby the murine LIS1 protein differs in only one aminoacid from the human version (Hirotsune et al. 1998; Reiner et al. 1993). In higher eukaryotes, LIS1 interacts with cytoplasmic dynein (NudA orthologue in A. nidulans) (Faulkner et al. 2000; Sasaki et al. 2000; Smith et al. 2000), which is critically involved in subcellular transport and directed cell movement (Kardon and Vale 2009). Lis1 interacts also with the two Nde1 and Ndel1 – formerly known a NudE and NudEL – paralogues that are homologous to A. nidulans NudE (Efimov and Morris 2000; Feng et al. 2000; Kitagawa et al. 2000; Niethammer et al. 2000; Sasaki et al. 2000; Smith et al. 2000). Structural analysis have revealed that both LIS1 and NDEL1 form homodimers (NDEL1 also tetramers) and that dimeric forms of NDEL1 are the principal configurations that interact with LIS1 (Bradshaw et al. 2009; Derewenda et al. 2007; Soares et al. 2012; Tarricone et al. 2004; Zylkiewicz et al. 2011). The interaction of LIS1 with NDE1 and NDEL1 critically influences the activity of cytoplasmic dynein (McKenney et al. 2010; Mesngon et al. 2006; Shmueli et al. 2010; Vallee et al. 2012; Yamada et al. 2008). In migrating cortical neurons, cytoplasmic dynein in concert with LIS1 act along microtubules to create forces, that eventually promote the saltatory nuclear movement toward the centrosome (Tsai and Gleeson 2005; Vallee et al. 2009). In fact, a recent model proposes that the Syne-1/2-SUN1/2 nuclear membrane proteins critically function in nucleokinesis by bridging the nucleus to the cytoskeleton via LIS1 and cytoplasmic dynein whereby the minus-end-directed microtubule motor dynein-dynactin complex then promotes nucleokinesis toward the centrosome (Zhang et al. 2009). This nucleus-centrosome coupling is the driving force and underlying molecular basis for the “two-stroke” model of neuronal migration (see also Chaps. 2, 4 and 7).
The regulation of the LIS1/NDEL1-complex and its functional interaction with dynein is controlled at multiple levels and it has been demonstrated that NDEL1 contains specific phosphorylation sites that are targeted by CDK5 (Niethammer et al. 2000) and Aurora A (Mori et al. 2007, 2009). NDEL1 is also regulated by palmitoylation on a conserved cysteine residue and it has been shown that palmitoylation of NDEL1 is essential for the function of cytoplasmic dynein and its downstream activities, including the control of cortical neuron migration (Shmueli et al. 2010). Besides posttranslational modifications regulating the LIS1/NDEL1-complex and thus dynein activity, NDEL1 (much more than LIS1) also serves as a structural and signaling platform for a large amount of interacting proteins [reviewed in (Chansard et al. 2011; Moon and Wynshaw-Boris 2013)].
3.2 Functional Analysis of the Lis1/Ndel1 in Cortical Neuron Migration In Vivo
The biochemical and biophysical studies described above resulted in a relatively detailed molecular model of how the LIS1/NDEL1-complex regulates dynein-mediated nuclear/somal migration (Tsai and Gleeson 2005; Vallee et al. 2009). In contrast, very little is known how the LIS1/NDEL1-complex orchestrates neuronal migration during cortical development in vivo. Lis1 and Ndel1 are essential genes and complete ablation of Lis1 and Ndel1 in knockout mice results in early embryonic lethality at E5.5 pre-implantation stages (Cahana et al. 2001; Hirotsune et al. 1998; Sasaki et al. 2005), and thus complicating the functional analysis of Lis1/Ndel1 function in vivo. Besides controlling neuronal migration, Lis1 has also essential functions in cell proliferation and neurogenesis which further compromises loss-of-function analysis of LIS1 during cortical neuron migration (Faulkner et al. 2000; Hippenmeyer et al. 2010; Tsai et al. 2005; Yingling et al. 2008). However, the dose of Lis1 is clearly very important and reduced levels of LIS1 in mice results in defects in the radial migration of several different types of neurons including cortical projection neurons (Cahana et al. 2001; Gambello et al. 2003; Hippenmeyer et al. 2010; Hirotsune et al. 1998). Systematic examination of the consequences of dosage reduction of LIS1 on cortical development was achieved by comparing wild-type (Lis1 +/+ ), null heterozygotes (Lis1 ko/+ ), compound heterozygote (null/hypomorph, Lis1 ko/hc ), and conditional null (Lis1 ko/hc ;Cre) and it was found that Lis1 likely controls the efficiency of neuronal migration in a dose-dependent manner (Cahana et al. 2001; Gambello et al. 2003; Hippenmeyer et al. 2010; Hirotsune et al. 1998; Youn et al. 2009). It remains unclear from these genetic in vivo studies whether Lis1 has specific functions at discrete steps in the radial migration process. In contrast, in utero electroporation of Lis1 small interference RNA and short hairpin dominant negative Lis1 in wild-type background in rat and mouse brains caused a dramatic accumulation of multipolar cells in the SVZ (Shu et al. 2004; Tsai et al. 2005). In addition, the above Lis1 knockdown experiments indicate that even bipolar neurons in the IZ critically depend on Lis1 function for locomotion, presumably by regulating nuclear translocation through centrosome-nucleus coupling (Shu et al. 2004; Tsai et al. 2005; Tsai and Gleeson 2005; Vallee et al. 2009). These findings are also in line with the total inhibition of migration phenotype observed in cortical slice assays upon complete loss of Lis1 in conditional null (Lis1 ko/hc ;Cre) (Youn et al. 2009). We have recently applied the MADM (Mosaic Analysis with Double Markers) strategy (Zong et al. 2005) to ablate Lis1 in sparse subpopulations of cortical projection neurons (Hippenmeyer et al. 2010). In the experimental MADM paradigm, homozygous mutant cells are labeled in one color (e.g. green by GFP), wild-type cells in another color (e.g. red by tdTomato) and heterozygous cells in yellow (i.e. green and red together) in an otherwise unlabeled background (Hippenmeyer et al. 2010; Zong et al. 2005). MADM-based functional gene analysis has the advantage that essential genes can be studied at any stage during development since the sparseness of gene knockout in single cells allows the bypass of embryonic lethality. Consequently, we have quantified the distribution of Lis1 +/+ , Lis1 +/− and Lis1 −/− cortical projection neurons at adult stages and noticed a significant reduction of Lis1 +/− and Lis1 −/− neurons in the uppermost layers II/III but concomitant increase in lower layers. Interestingly, there was no significant difference between Lis1 +/− and Lis1 −/− in their laminar distribution although the migration of Lis1 −/− neurons is (at least initially) significantly delayed at birth when compared to Lis1 +/− . In contrast, Lis1 +/+ neurons displayed a significant cell-autonomous migration advantage when compared to Lis1 +/− and Lis1 −/− neurons, respectively. The presence of MADM-labeled Lis1 −/− neurons in all cortical layers indicates that non-autonomous and/or community effects could strongly influence the positioning of these Lis1 −/− mutant (and heterozygous Lis1 +/− ) neurons (see also below), given the critical function of Lis1 in promoting nuclear/somal translocation. Future detailed MADM-based analysis of Lis1 at embryonic stages may promise further insight into the cell-autonomous and possibly cell-nonautonomous functions of Lis1 in regulating the sequential steps of cortical neuron migration. In summary, Lis1 acts in a dose-sensitive manner to regulate the efficiency of neuronal migration, likely by promoting somal translocation via nucleus-centrosome coupling in migratory neurons and catalyzes the exit from the multipolar conformation to the bipolar state of nascent neurons in the embryonic SVZ (Fig. 1.3a). Experiments involving mosaic ablation of Lis1 by MADM in overall heterozygote Lis1 +/− mice also indicate a significant degree of intersecting community effects (upon reduction of Lis1) and/or cell-nonautonomous function(s) for Lis1 in regulating neuronal migration (Hippenmeyer et al. 2010; Youn et al. 2009).
Fig. 1.3
Cell-autonomous and non-autonomous functions of Lis1 / Ndel1 at specific steps during radial migration. Models of cell-autonomous (green) and non-autonomous (blue) in vivo functions of LIS1 (a) and NDEL1 (b) in the developing cortex. LIS1 cell-autonomously regulates the efficiency of neuronal migration in a dose-dependent manner and has a role in the multi-to-bipolar transition of cortical projection neurons. NDEL1 cell-autonomously controls invasion and/or migration within the developing CP target laminae. Extensive interactions among migrating neurons, either mediated by specific cell-nonautonomous effects of LIS1/NDEL1 or through a general community effect, promote migration of Ndel1 −/− cells before reaching the target laminae and Lis1 −/− cells along the entire path under sparse knockout conditions (Adapted from (Hippenmeyer et al. 2010) with permission)
Like for Lis1, homozygous complete knockout of Ndel1 in mice results in embryonic lethality (Sasaki et al. 2005) and although heterozygote Ndel1 +/− mice do not show obvious phenotypes, further reduction of Ndel1 in Ndel1 ko/hc (null/hypomorph) animals results in mild neuronal migration phenotypes (Sasaki et al. 2005; Youn et al. 2009). Ndel1 RNAi knockdown experiments by in utero electroporation independently revealed an essential function for Ndel1 for appropriate cortical projection neuron migration (Shu et al. 2004). The function of Ndel1 involves proper coupling of the centrosome to the nucleus, presumably in a Lis1 dependent manner since the requirement for Ndel1 in cortical neuron migration could be partially compensated by overexpression of Lis1 (Shu et al. 2004). Complete loss of Ndel1 in conditional knockout mice (Ndel1 ko/hc ;Cre) led to total inhibition of neuronal migration in organotypic cortical brain slices as revealed by high resolution time-lapse two-photon videomicroscopy (Youn et al. 2009). However, by using the same migration assay, reduction of NDEL1 protein levels to about 35 % in Ndel1 ko/hc allowed these neurons to migrate, albeit at reduced speeds. The slower migrating neurons in Ndel1 ko/hc displayed multiple branches, like multipolar cells, and exhibited a branched migration pattern (Youn et al. 2009). Such branched migration is very similar to the movements of neurons lacking p35 (Chae et al. 1997; Gupta et al. 2003), which is the activator of CDK5 (Tsai et al. 1994). In p35 −/− mutant mice (Chae et al. 1997) the vast majority of radially migrating cortical projection neurons displayed branched migration behavior (Gupta et al. 2003). Importantly, p35/CDK5 phosphorylates NDEL1 at specific sites (S198, T219 and S231) and this phosphorylation is essential for NDEL1 function (Niethammer et al. 2000; Sasaki et al. 2000). Therefore it is likely that the similar neuronal migration phenotypes of neurons lacking p35 or having reduced levels of NDEL1 could be causally related. Whether p35/CDK5-mediated NDEL1 phosphorylation is critically required during the entire radial neuron migration process or just at discrete steps is an important open question. In fact, loss of CDK5 function results in the accumulation migrating cortical projection neurons in the IZ and many of these Cdk5 −/− mutant neurons remain in a multipolar configuration (Gilmore et al. 1998; Ohshima et al. 1996, 2007). Thus, p35/CDK5-induced phosphorylation of NDEL1 could in principle, besides promoting migration, trigger the multi-to-bipolar transition although distinct pathways including other CDK5 target molecules likely act in parallel in these sequential radial migration steps.
In order to get further insights and identify the critical step(s) in cortical neuron migration controlled by NDEL1 function in vivo, we pursued functional MADM analysis of Ndel1 (Hippenmeyer et al. 2010). By using developmental time course and single clone analyses, the phenotype of mutant Ndel1 −/− MADM-labeled cells revealed that Ndel1 cell-autonomously regulates a very specific step in cortical neuron migration: the entry into the CP (Fig. 1.3b). Live-imaging experiments of MADM-lableled neurons in organotypic slice preparations further indicated that the speed of migration for Ndel1 +/+ and Ndel1 −/− neurons is not significantly different as long as the neurons migrate within the IZ. In contrast, the majority of Ndel1 −/− neurons failed to cross the IZ-CP border (Fig. 1.4). Altogether, the MADM experiments showed that Ndel1 −/− neurons in a mosaic, mostly heterozygous, environment can migrate through the VZ/SVZ and the entire IZ. However, in the MADM setting, Ndel1 −/− neurons are unable to enter and/or migrate into their CP target lamina. The analysis of several different types of MADM-labeled Ndel1 −/− mutant neurons, besides cortical projection neurons, indicates that NDEL1 is generally required for the invasion of the target area in the course of radial neuronal migration (Hippenmeyer et al. 2010). The mosaic analyses afforded by MADM not only revealed cell-autonomous functions of Ndel1 (and Lis1, see above) but indicates a significant amount of cell-nonautonomous effects which critically contribute to the regulation of discrete steps in radial neuron migration. Most indicative for such cell-nonautonomous effects is the fact that the phenotypes resulting from sparse MADM-based gene knockout are distinct from those observed in whole cortex knockout (Hippenmeyer et al. 2010; Youn et al. 2009) (Fig. 1.4). For instance, Ndel1 −/− mutant neurons were unable to move in cultured organotypic brain slices from Ndel1 ko/hc ;Cre mice (complete loss of NDEL1 in cortex) but MADM-labeled Ndel1 −/− neurons (in genetic mosaic, mostly normal environment) could migrate at regular speeds in IZ and only stalled once they reached their CP target area. Thus, it is conceivable that the inability of Ndel1 −/− mutant neurons to migrate in Ndel1 ko/hc ;Cre mice is potentially a consequence of adversive cell-nonautonomous effects. What could be the molecular nature of such ‘negative’ effects? Neuronal migration requires the dynamic adjustment of cell adhesion molecules such as N-cadherin (see also below) and possibly axon guidance molecules (see also Chap. 9). Thus, one could speculate that the overall molecular landscape at the cell membrane might be non-permissive for migration and/or even inhibiting when Ndel1 −/− neurons contact each other. Conversely, in a mosaic MADM environment, most Ndel1 −/− mutant neurons only get in contact with Ndel1 +/− or Ndel1 +/+ neurons and cross-inhibiting Ndel1 −/− – Ndel1 −/− cell contacts are minimized. On the other hand, migrating MADM-labeled Ndel1 −/− neurons, as long as progressing through the VZ/SVZ and the IZ, could even be positively influenced by the neighboring Ndel1 +/− or Ndel1 +/+ neurons. Such ‘positive’ effects might also account for the positioning of MADM-labeled Lis1 −/− in upper cortical layers (see above). The precise nature of the ‘positive’ cell-nonautonomous effects and how they account for positioning and/or migration of mutant Ndel1 −/− and Lis1 −/− cortical projection neurons is currently unclear. In principle two scenarios could be possible: (1) community effects whereby isolated Ndel1 −/− or Lis1 −/− neurons, despite defective in the intrinsic cytoskeletal migration machinery (e.g. nucleus-centrosome coupling), may still “piggyback” on normally migrating abutting neurons; and (2) neurons migrating in a crowd could actively signal, in a Lis1/Ndel1-dependent manner, to each other in order to boost the intrinsic migration machinery. Molecularly, such a mechanism poses however a significant challenge since intracellular cytoplasmic proteins would need to signal inside-out and trans-cellular to neighboring migrating cells. Such a signaling pathway likely would need to involve transmembrane receptors and possibly extracellular matrix components, and some sort of a sensor connecting to cytoskeletal force generators in the ‘signal-receiving’ cell. Interestingly, cell-on-cell migration of Drosophila invasive border cells, which also depends on cell-autonomous requirement for dLis1, has been proposed to involve “piggy-backing” (Yang et al. 2012). More generally, community effects have been observed in a variety of other cell types moving collectively (Friedl and Gilmour 2009; Tada and Heisenberg 2012; Theveneau and Mayor 2013).
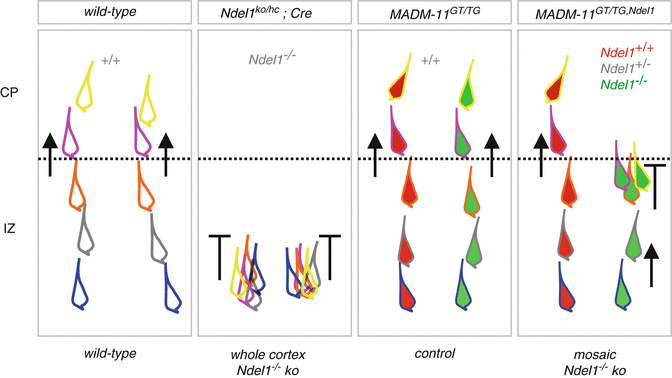
Fig. 1.4
Migration phenotypes of Ndel1 −/− cortical projection neurons in whole and sparse knockout experimental paradigms. Distinct migration phenotypes in the IZ and CP in wild-type, Ndel1 ko/hc ;Cre (whole cortex Ndel1 −/− knockout), MADM-11 GT/TG (control, all cells Ndel1 +/+ ) and MADM-11 GT/TG,Ndel1 (mosaic Ndel1 −/− knockout). The different colored outlines of the migrating neurons shall indicate sequential positions at distinct times during progressive radial migration. This summary is based on the experimental time-lapse imaging data from (Hippenmeyer et al. 2010; Youn et al. 2009)
The adaptor protein 14-3-3ε is encoded by YWHAE in human and deleted in all MDS patients (Cardoso et al. 2003; Toyo-oka et al. 2003; Wynshaw-Boris 2007). The precise function of 14-3-3ε during brain development is unclear but it has been shown that Ywhae genetically interacts with Lis1 in mice and that the 14-3-3ε protein binds to p35/CDK5 phosphorylated NDEL1 (Toyo-oka et al. 2003). The 14-3-3ε/NDEL1 interaction is important for binding to LIS1 and the dynein motor. Thus, the tripartite LIS1/NDEL1/14-3-3ε complex appears as a key regulator of neuronal migration. Interestingly, sparse MADM-mediated conditional knockout of Ywhae in cortical projection neurons did not significantly affect their radial migration although Ywhae −/− pyramidal CA1 cells in hippocampus showed slight defects in migration (Hippenmeyer et al. 2010). Since multiple isoforms of 14-3-3 are expressed in the brain (Takahashi 2003), distinct 14-3-3 isoforms could compensate for the loss of 14-3-3ε function. Along the same lines, genetic redundancy could also, at least in part, explain some of the divergent phenotypes that were observed when comparing the individual MADM-induced Lis1 −/− , Ndel1 −/− and 14-3-3ε −/− mutant neurons to each other. For instance Nde1, the second homologue of A. nidulans NudE and paralogue of murine Ndel1, shares 55 % sequence homology with Ndel1 (Feng et al. 2000; Sweeney et al. 2001) and could possibly compensate for some Ndel1 functions, especially during the early stages of neuronal migration in VZ/SVZ and IZ. Although the overall phenotype of Nde1 −/− mutant mice is distinct from Ndel1 −/− mutants, NDE1 can interact with LIS1 and this interaction is critical to control cortical lamination and thus cortical neuron migration (Feng et al. 2000; Feng and Walsh 2004; Pawlisz et al. 2008). Alternatively, LIS1, NDEL1 and 14-3-3ε may also each act in a ‘complex’-independent manner to execute distinct component-specific functions, at discrete steps and transitions during radial migration of cortical projection neurons. Recent biochemical experiments support such a model whereby for example both LIS1 and NDEL1 proteins may act independently in certain contexts and that their interaction can amplify and/or modulate those activities (Zylkiewicz et al. 2011).
4 The Reelin Pathway Controls Sequential Phases in Cortical Neuron Migration
Cortical layering occurs in an ‘inside-out’ fashion whereby earlier born neurons populate deep layers and later born neurons occupy progressively upper layers (Angevine and Sidman 1961; McConnell 1995; Polleux et al. 1997; Rakic 1974, 2007). The Reelin signaling pathway plays fundamental roles in cortical neuron migration and inside-out lamination, and has attracted unmatched attention, by now for over half a decade. In human, mutations in the RELN gene (encoding RELN also known as Reelin) are associated with autosomal recessive lissencephaly and cerebellar hypoplasia (Hong et al. 2000) but the precise function and mode of action of the Reelin signaling cascade at the cellular and molecular level in vivo remains unclear.
Historically, Falconer initially described the Reeler mouse mutant phenotype which includes ataxia, tremors and a reeling gait (Falconer 1951), hence the name Reeler. A multitude of anatomical studies has been carried out but the most striking feature of the Reeler mouse is that laminated brain structures including the neocortex are disorganized with misplaced projection neurons (Caviness and Rakic 1978; Hamburgh 1963; Tissir and Goffinet 2003). Upon cloning of the Reelin gene Reln in mouse it became evident that Reelin encodes a large (3,461 amino acids) secreted glycoprotein (D’Arcangelo et al. 1995). Reelin is mostly expressed and secreted from the earliest born cortical ‘pioneer’ Cajal-Retzius cells which form the outermost layer of the PP and later the MZ (D’Arcangelo et al. 1995; Ogawa et al. 1995). Reelin binds to a receptor complex – VLDLR (very low-density lipoprotein receptor)/LRP8 (low-density lipoprotein related receptor 8, formerly known as APOER2) (D’Arcangelo et al. 1999; Hiesberger et al. 1999) and strikingly, the phenotype of Vldlr/Lrp8 double knockout mice is basically indistinguishable from the Reeler phenotype (Trommsdorff et al. 1999). The Reelin signal is transmitted via the intracellular signaling adaptor DAB1 which is phosphorylated by Src-family kinases upon Reelin binding (Arnaud et al. 2003; Bock and Herz 2003; Howell et al. 1999), and Dab1 mutant mice (Howell et al. 1997; Sheldon et al. 1997; Ware et al. 1997) exhibit neurological and anatomical phenotypes the are indistinguishable from the ones in Reeler mutant mice (Trommsdorff et al. 1999). Both VLDLR and LRP8 do not possess intrinsic kinase activity and it has been shown that the axon guidance molecule ephrin-B can act as a co-receptor for Reelin (Senturk et al. 2011). Reelin binds to the extracellular domain of ephrin-Bs and thereby associating with the VLDLR/LRP8 complex. This leads to clustering of ephrin-Bs and Src kinase-mediated DAB1 phosphorylation and activation (Senturk et al. 2011). Finally, Notch signaling, executing most important functions during neurogenesis (Kageyama et al. 2009) has recently been shown to play a critical role in transducing the Reelin signal during neuronal migration (Hashimoto-Torii et al. 2008) although the precise mechanisms how Notch functions as a Reelin downstream mediator still remain enigmatic.
Despite tremendous efforts to decipher the biological Reelin function(s) in cortical neuron migration, important aspects are still unclear and several somewhat conflicting hypotheses have been put forth. It has been proposed that Reelin might act (1) as a repellent cue (Ogawa et al. 1995; Schiffmann et al. 1997); (2) as a stop signal (Frotscher 1997; Sheppard and Pearlman 1997); (3) to stimulate detachment of migrating cortical neurons from the radial glia process (Dulabon et al. 2000; Sanada et al. 2004) or (4) specifically could regulate radial glia-independent somal translocation (Franco et al. 2011; Jossin and Cooper 2011; Sekine et al. 2012) (see also below).
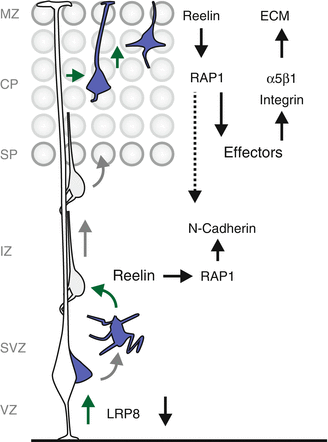
One reason for the apparent pleiotropy of the Reelin deficiency phenotypes could be that besides cell-autonomous functions of Reelin signaling in migrating neurons, environmental factors, community and other cell-nonautonomous effects might critically influence Reelin-sensitive neurons (Franco et al. 2011; Sanada et al. 2004). In any case, several recent experiments convincingly suggest that Reelin may play not only one but instead several important cell-autonomous roles and likely acts at multiple discrete steps during radial migration of cortical projection neurons. First, Reelin-dependent LRP8 downregulation could be important for nascent neurons to delaminate and start their migration by uncoupling them from neural progenitor cells (Perez-Martinez et al. 2012). It is however not clear if this proposed early function acts more or less as a permissive signal rather than acting as an instructive cue. Second, Reelin plays an essential role in the multipolar-to-bipolar transition while neurons migrate within the IZ (Jossin 2011; Jossin and Cooper 2011). In fact, migrating neurons in the IZ show the highest level of “functional Reelin receptors” and the Reelin signal could diffuse from the MZ to the IZ (Jossin et al. 2007; Uchida et al. 2009). Jossin and Cooper propose a three step model how Reelin regulates the multipolar-to-bipolar transition of cortical neurons in the IZ. In a first step, multipolar neurons migrate in an apparently stochastic mode in the IZ where they encounter Reelin, which leads to activation of the small GTPase RAP1, presumably via pDAB1-CRK/CRKL-C3G signaling (Ballif et al. 2004; Voss et al. 2008). RAP1 is a Ras-related GTPase which plays important roles in the regulation of the actin cytoskeleton, membrane trafficking and cell adhesion (Gloerich and Bos 2011). In the second step, active RAP1 increases the surface level of N-Cadherin in the multipolar neurons. The increased levels of surface N-Cadherin could then allow the multipolar neurons to sense environmental cues allowing the proper polarization and exit of the multipolar state to adopt bipolar morphology (Jossin 2011). While the precise intracellular signaling mechanisms controlling RAP1-mediated N-Cadherin regulation during multi-to-bipolar transition and migration towards the upper IZ remain to be elucidated, Reelin also regulates later aspects in the radial migration process. Upon transition to the bipolar morphology, cortical neurons migrate along the radial glia processes towards the cortical plate. This glia-dependent migration (locomotion) mode appears to be independent of Reelin signaling. In contrast, the very last step – terminal somal translocation – which seems to occur in a glia-independent manner requires DAB1-mediated Reelin signaling (Franco et al. 2011; Jossin and Cooper 2011; Sekine et al. 2012). Interestingly, such glia-independent somal translocation is the predominant mode of migration of early born neurons occupying the future lower cortical layers (Nadarajah and Parnavelas 2002). Thus DAB1-mediated Reelin signaling likely promotes cell-autonomously the splitting of the PP by deep layer neurons.
Because early and late born neurons travel different distances and through varying cellular compartments in their individual migration journeys, it is likely that the molecular machineries driving their migration could be different for future deep versus upper layer neurons, respectively. Therefore, neurons destined for different layers most likely have additional distinct requirements for functional Reelin signaling. Nevertheless, for the very last step in the migration process – terminal somal translocation – which is conserved in early and late born neurons, Reelin is equally important in both early and late born neurons (Franco et al. 2011; Jossin and Cooper 2011; Sekine et al. 2012). Mechanistically, Reelin signaling via RAP1 and subsequent control of N-cadherin function seems to promote terminal somal translocation (Franco et al. 2011; Jossin and Cooper 2011). The functions of N-cadherin in the control of radial neuron migration appear however pleiotropic since also glia-dependent locomotion depends on N-cadherin function (Kawauchi et al. 2010). How N-cadherin signaling – triggered either by Reelin-RAP1-mediated intracellular redistribution (multi-to-bipolar transition and glia-independent terminal somal translocation) or via Reelin-independent pathways (glia-dependent locomotion) – regulates the distinct sequential steps of neuronal migration is currently not well understood. Precise regulation of N-cadherin levels through endocytic and/or coordinated intracellular trafficking pathways could be an effective way for appropriately tuning of N-cadherin levels/activity (Kawauchi et al. 2010). As such, during the locomotion process of migrating neurons, N-cadherin undergoes RAB5-dependent endocytosis at the trailing end and is efficiently shuffled to the plasma membrane at the forward end via a RAB11-dependent recycling pathway. Conversely, RAB7-dependent lysosomal degradation pathways with increased degradation of N-cadherin and possibly other cell adhesion molecules seem also important for the final terminal somal translocation step (Kawauchi et al. 2010). Although the Reelin-DAB1-CRK/CRKL-C3G-RAP1 pathway dynamically regulates N-cadherin activity and terminal somal translocation, overexpression of N-cadherin alone was not sufficient to rescue loss of Reelin-mediated DAB1 signaling (Franco et al. 2011). Therefore, distinct signaling molecules and/or cell adhesion molecules have additional roles in the transmission of the Reelin signal for the promotion of terminal somal translocation. Consequently, it has been demonstrated that Reelin triggers C3G-RAP1-mediated integrin activation in an inside-out signaling fashion specifically during somal translocation (Sekine et al. 2012). In this pathway, Reelin acts as input signal through VLDLR/LRP8-DAB1-CRK/CRKL-C3G to activate RAP1 which recruits effectors to promote conformational change and thus activation of α5β1-integrin, which then triggers adhesion to fibronectin in the ECM of the MZ to mediate terminal somal translocation (Sekine et al. 2012).
Altogether and despite tremendous efforts and progress in deciphering the biological functions of the Reelin signaling pathway, the precise mechanism of the highly complex Reelin signaling cue and how it regulates the proper positioning of neurons in the CP remains unclear and somewhat controversial. There is an emerging picture whereby Reelin controls radial neuronal migration at several distinct steps (Fig. 1.5). Reelin acts (1) during delamination and separation of nascent neurons from neuronal progenitors; (2) to regulate the multi-to-bipolar transition; and (3) most importantly during somal translocation of early born lower layer neurons and terminal somal translocation of late born upper layer neurons. Interestingly, the Reelin and LIS1 pathways described above are likely to intersect at certain stages during cortical neuron migration (Assadi et al. 2003). However, the cytoarchitectural phenotypes of the cortex in mice with absent Reelin or LIS1 are distinct (Caviness and Rakic 1978; D’Arcangelo et al. 1995; Hirotsune et al. 1998; Yingling et al. 2008) although loss of function of either pathway cause lissencephaly in human (Hong et al. 2000; Reiner et al. 1993). At which steps during radial migration the Reelin and LIS1 signaling pathways crosstalk to exert their critical function(s) in driving migrating cortical neurons awaits further investigation.
 The Impact of JNK on Neuronal Migration
The Dynamics of Neuronal Migration
Spinal Motor Neuron Migration and the Significance of Topographic Organization in the Nervous System
Mark/Par-1 Marking the Polarity of Migrating Neurons
The PAR Polarity Complex and Cerebellar Granule Neuron Migration
Extracellular Signals Controlling Neuroblast Migration in the Postnatal Brain
The Impact of JNK on Neuronal Migration
The Dynamics of Neuronal Migration
Spinal Motor Neuron Migration and the Significance of Topographic Organization in the Nervous System
Mark/Par-1 Marking the Polarity of Migrating Neurons
The PAR Polarity Complex and Cerebellar Granule Neuron Migration
Extracellular Signals Controlling Neuroblast Migration in the Postnatal Brain




Related posts:
The Impact of JNK on Neuronal Migration
The Dynamics of Neuronal Migration
Spinal Motor Neuron Migration and the Significance of Topographic Organization in the Nervous System
Mark/Par-1 Marking the Polarity of Migrating Neurons
The PAR Polarity Complex and Cerebellar Granule Neuron Migration
Extracellular Signals Controlling Neuroblast Migration in the Postnatal Brain
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
