Fig. 3.1
Scheme of the JNK cascade and the upstream components that regulate cell migration during cortical development. (a) JNKs are the effector kinases that reside at the end of a classical MAPK signalling cascade. Studies in transgenic mice have indicated a role for DLK and MEKK4 in the regulation of neuronal migration, and for MEKK1 in the regulation of epithelial cell migration during eye development. MEKK4 signals via p38 to positively regulate neuronal migration (green arrow), in contrast JNK1 slows (red arrow) the rate of multipolar and bipolar cell migration. The MAPK splice variants involved in migration regulation are not known, though multiple forms are expressed in brain. (b) Depicted in the scheme are those receptors that activate JNK in neuronal systems in various contexts and may contribute to JNK regulation during neuronal migration. ApoER2 is the Reelin receptor that couples to JNK (Trommsdorff et al. 1999), BMP7 binding to BMPRII and activates JNK (Podkowa et al. 2010), endothelin-1 activates JNK via the ETB receptor in developing brain (Mizuno et al. 2005). NMDA receptor activation also induces JNK activity in neurons (Mukherjee et al. 1999)
2 JNKs Regulate Cell Death in Developing and Ageing Brain
In non-neuronal cells JNK activity is low and triggered in response to stressful stimuli. However, in primary neuron cultures and in brain tissue JNK activity is anomalously high even in the absence of stress (Coffey et al. 2000; Coffey and Courtney 1997; Hu et al. 1997). This elevated activity in brain derives mainly from JNK1 isoforms as 50 % of resting JNK activity is lost in the cortex of Jnk1 −/− mice, while JNK2 and JNK3 isoforms contribute less (Tararuk et al. 2006). Precise measurements of stress responsive JNK in neuronal systems are for this reason compounded by contaminating constitutive kinase activity. However, JNK2/3 isoforms have been isolated using isoform-specific antibodies and shown to be activated by stress in the trophic factor deprivation model of developmental death in neurons (Coffey et al. 2002). JNK3 has been implicated in excitotoxic damage in brain and Jnk3 −/− adult mice show reduced sensitivity to kainate-induced seizures (Yang et al. 1997b; Kuan et al. 2003; Pirianov et al. 2007) while Jnk3 −/− neonates are less susceptible to hypoxia-ischemia induced damage (Pirianov et al. 2007). Furthermore, cortical and hippocampal neurons derived from Jnk3 −/− mice are protected from beta-amyloid-induced death (Morishima et al. 2001). In the peripheral nervous system, Jnk3 −/− neurons are protected from axotomy-induced death and JNK pathway inhibitors protect in models of Parkinson’s disease (Keramaris et al. 2005; Maroney et al. 1999; Brecht et al. 2005). Studies evaluating neuronal death during development, have shown that all JNKs (1, 2 and 3) contribute to toxicity, and significant protection requires silencing of all three genes (Björkblom et al. 2008). Moreover, the JNK substrates mediating neuronal death are not yet formally identified. While increased phosphorylation of JNK substrates c-Jun and ATF2 accompanies JNK activation in a range of neuronal death models, efficient knockdown of c-Jun or Atf2 in neurons does not protect from death (Björkblom et al. 2008), and c-Jun phosphorylation was unchanged in Jnk1/2 −/− double mutants suggesting that c-Jun phosphorylation by JNK is not essential for brain morphogenesis and the accompanying apoptosis changes (Behrens et al. 1999; Sabapathy et al. 1999). Although it is not easy to formulate a simple model to explain how JNKs contribute to both pathological and physiological functions, for example based on isoform dependence alone, the signaling complexity does not take away from the wealth of evidence that indicates that these kinases are major effectors of cell death in the nervous system. Indeed, inhibition of JNK activity is elevated in post-mortem brain from patients with Parkinson’s Disease and Dementia with Lewy bodies and in Alzheimer’s patients (Ferrer et al. 2001, 2002), and inhibition of JNK provides neuroprotection in an extensive range of neuronal death models (Waetzig et al. 2006).
3 Developmental Expression in the Nervous System
Jnk1 and Jnk2 are expressed ubiquitously throughout the body including brain, while Jnk3 expression is almost exclusively expressed in brain with low levels found in heart and testis (Kuan et al. 1999). Jnk1, 2 and 3 mRNAs are high in embryonic brain (Kuan et al. 1999) and in adult brain JNK1 protein expression is elevated in the cortex, hippocampus and striatum while JNK3 expression is high in the hippocampus, lower levels are detected in the cortex, striatum and cerebellum (Brecht et al. 2005). Directly upstream from JNK, Mkk4 and Mkk7 mRNAs are ubiquitously expressed in adult mouse brain, whereas in the developing nervous system, Mkk4 appears before E10 after which it is also expressed in liver and thymus (Wang et al. 2007c). As development proceeds MKK4 protein levels increase in brain, reaching a stable level in adulthood, while MKK4 simultaneously decreases in the liver and thymus (Lee et al. 1999). In contrast, MKK7 shows wide tissue expression both in during development and in adult. However at later stages of embryogenesis MKK7 levels in brain, skin and hair follicle increase (Yao et al. 1997).
4 Lessons Learned from Genetic Disruption of JNKs and Upstream Kinases in Mice
4.1 Jnk1 and Jnk2 Regulate Early Brain Morphogenesis
Genetic ablation of Jnk1 and Jnk2 in mice results in embryonic death at around E11.5 with defective neural tube closure (Kuan et al. 1999). These mice show regional specific apoptosis with decreased cell death in the hindbrain neuroepithelium just prior to neural tube closure, and increased apoptosis and caspase-3 activation in the forebrain (Sabapathy et al. 1999; Kuan et al. 1999). Neural tube closure is a process that requires cell migration, cell death and cell proliferation. However, major defects in neural crest cell migration were not described in Jnk1/2 −/− double mutants (Kuan et al. 1999; Sabapathy et al. 1999). Similarly the number of BrdU positive cells (reflecting the proliferating population), were unchanged and the exencephaly phenotype (where the brain is completely exposed or protrudes outside of the skull) was therefore attributed to deregulated apoptosis. The open neural tube phenotype showed a gene dosage effect since Jnk1 −/− ;Jnk2 +/− mice displayed exencephaly, while Jnk1 +/− ;Jnk2 −/− mice did not (Sabapathy et al. 1999), indicating that JNK1 plays a dominant role in regulating the events underlying neural tube closure (Sabapathy et al. 1999). These studies marked a turning point in the field of JNK research as they established an important role for JNK function in the developing nervous system and demonstrated a critical role in regulation of cranial morphogenesis. Subsequent work has shown that JNKs are fundamentally important for migration of several cell types including epithelial cells (Huang et al. 2004; Yamasaki et al. 2012). Therefore, the possibility that migration defects contribute to the exencephaly phenotype of Jnk1/2 −/− double mutant mice cannot be excluded. Indeed these studies focused on quantitative measures of apoptosis and qualitative measures of cell proliferation. A more detailed analysis of cell migration in Jnk1/2 −/− double mutants during early embryogenesis may therefore be worth a re-visit.
It is notable that the classic phenotypic neural tube defect observed in Jnk1/2 −/− double mutant mice is not replicated by genetic ablation of either of the upstream regulators Mkk4 or Mkk7 (Table 3.1) (Wang et al. 2007b, c; Wada et al. 2004), while double knockout mice die at E8.5 prior to neural tube closure. Thus the activation of JNK, in the context of neural tube closure, may require a coordinated regulation by both MKK4 and MKK7, or alternatively may involve an unknown mechanism.
Table 3.1
Summary of the major phenotypes in Jnk and Map2k transgenic mice
Jnk1 −/− | Jnk2 −/− | Jnk3 −/− | Jnk1 −/− ;Jnk2 −/− | Mkk4 −/− | Mkk4 flox/flox ; Nestin-Cre | Mkk7 −/− | Mkk7 flox/flox ; Nestin-Cre | ||
|---|---|---|---|---|---|---|---|---|---|
Phenotype | Viable. Disorganized neuron positioning in cortex, increased multipolar and bipolar motility ratea. Decreased duration of multipolar stagea. | Viable. No overt brain malformations | Viable. No overt brain malformations | E11.5 lethal | Die E11.5-E13.5 from anemia and abnormal hepatogenesisg | Irregular Purkinje cell positioning and delayed radial migrationi | E 11.5-E12.5 lethalj | Enlarged ventricles by E18.5. Diminished striatum, disturbed development of axonal tractsl | |
Impaired neural tube closured | Degeneration of the anterior commissure in adult mousei | Reduced neurite length at E15.5l Altered radial migration l (Yamasaki et al. 2011) | |||||||
Anterior commissure lost by 3 monthsb | |||||||||
Altered dendrite morphologyb,c | |||||||||
Cell death proliferation | A small decrease in cell cycle exit in embryonic braina | Resistance to MPTP toxicitye | Resistance to MPTP and kainate induced neurotoxicity and seizurese,f | Increased death in hindbrain, decreased death in forebrain at E11.5d | Increased hepatocyte apoptosis and liver degenerationg | Decreased cell proliferation and defective liver formation with reduced parenchymal hepatocyte numberj | Increased presence of autophagic vacuolesl | ||
Tissue expression profile of respective MAPK/MAP2K | Jnk1 from E7 onwards in different tissues including brain, heart, liver, lungd | Jnk2 E7 onwards in brain, heart, lung, liverd | Jnk3 E11 onwards in brain and lower levels in heart and testisd | Jnk1/Jnk2 E7 onwards in various tissues including brain, heart, liver, lungd | Mkk4 expressed by E10 in CNS. E12 onwards, transcripts in liver, heart, immune systemh | Mkk7 is present in embryonic brain in epithelial tissuesk |
The JNK substrates contributing to cranial morphogenesis have not been clarified though transcription factor targets of JNK could be central. Mice null for the JNK substrates c-Jun or Atf2 do not display defects in cranial morphogenesis indicating that they either play redundant roles in this process or none at all. Interestingly, c-Jun −/− mice do share the Mkk4 and Mkk7 phenotype of deregulated hepatocyte proliferation, while Atf2 −/− mice die at birth with respiratory defects (Eferl et al. 1999; Maekawa et al. 1999). More recently, a screen for novel JNK substrates in brain revealed an actin regulatory protein (required for neural tube formation) which is myristoylated Alanine Rich C-Kinase Substrate Like protein-1 (MARCKSL1), also known as F52, Mac MARCKS and MRP (Björkblom et al. 2012). MARCKSL1 is phosphorylated on three sites by JNK and genetic deletion of MARCKSL1 results in exencephaly (Wu et al. 1996; Björkblom et al. 2012). MARCKSL1 is the only known JNK substrate to date that phenocopies Jnk1/2 −/− double mutant mice.
4.2 JNK1 Regulates Multipolar Stage Exit and the Rate of Movement of Multipolar and Bipolar Cells in Developing Cortex
Movement of cells from their place of birth to their final destination is a fundamental feature of brain morphogenesis. The speed at which neurons move to form the layers of the cortex could affect their final placement. The role of JNK in regulating the rate of nerve cell movement has been examined in the context of multipolar cell migration and bipolar cell locomotion and is discussed in the following paragraph. The multipolar phase occurs when the precursors of pyramidal neurons in the intermediate (IZ) and subventricular (SVZ) zones take on a multipolar shape and display a movement that is characterized by frequent changes in direction and a slower speed compared to locomoting cells moving on radial glia processes (Tabata and Nakajima 2003). The molecular mechanisms of multipolar migration and multipolar-to-bipolar transition have only recently received attention, as described in more detail below (Westerlund et al. 2011; Tabata et al. 2009; Pacary et al. 2011). At the level of gene regulation, transcription factors determining glutamatergic neuron differentiation are known as gene regulators of multipolar transition. Recently a role for the transcriptional repressor RP58 governing multipolar-to-bipolar conversion has been described. RP58 represses Ngn2 transcription and thereby regulates the Ngn2-Rnd2 pathway (Geisen et al. 2008; Nóbrega-Pereira et al. 2008; Ohtaka-Maruyama et al. 2013) (see also Chap. 1).
JNK1 is directly implicated in regulation of multipolar phase exit and neuronal migration during cortical development (Westerlund et al. 2011). This study used in utero electroporation to label neuroepithelial progenitors at E15.5 and demonstrated that GFP-expressing neurons in Jnk1 −/− mice migrated faster than those in wild-type mice. Ex vivo imaging in organotypic slices demonstrated a bipolar cell movement rate of 5–10 μm per hour in wild-type compared to 15–20 μm per hour in Jnk1 −/− cortex. The multipolar cells also moved at higher speeds and transit more quickly to the bipolar phase with subsequent pial-directed migration (Westerlund et al. 2011). This was illustrated by a relatively large ratio of bipolar to multipolar cells in the IZ and reduced duration of the multipolar phase. Furthermore, the JNK binding domain (JBD) of JIP [JIP1a(1–277)], a peptide inhibitor of JNK that binds tightly to JNK and prevents substrate binding (Dickens et al. 1997), was used in cortical neurons. Neurons expressing GFP-JBD in vivo reached the cortical plate (CP) faster than in control mice (Mizuno et al. 2005; Westerlund et al. 2011). Despite the altered migration speed, no overt lamination defect was detected in Jnk1 −/− mice, in contrast to Cdk5 −/− or p35 −/− mice that exhibit disturbed layering (Gilmore et al. 1998; Gupta et al. 2003). However, in Jnk1 −/− mice, the final cell positioning was quite markedly disorganized and crowded (Westerlund et al. 2011). This mal-positioning may be a consequence of the unusually rapid transit through the multipolar phase, when it is most probable that choices are made to influence the final cellular placement. This could include selection of the appropriate radial glial scaffold for instance. The findings from Jnk1 −/− mice compare strikingly well to those obtained with Reeler mice (Britto et al. 2011), and are discussed in more detail later in this chapter. These results thus suggest that JNK1 may be a conveyor of Reelin’s directive on cell positioning in the developing cortex.
While it is not clear whether JNK2 or JNK3 regulate neuronal migration, it is worth noting that opposing results were obtained using the cytosolic or nuclear-targeted JBD inhibitor. Inhibition of JNK in the cytoplasm using compartment specific inhibitors (Björkblom et al. 2005; Tararuk et al. 2006) led to an accelerated migration phenotype, thereby mimicking Jnk1 −/− mice, while inhibition of nuclear JNK had the opposite effect (Westerlund et al. 2011). These results suggest that in addition to cytosolic targets, JNK controls a gene transcription program that is required for migrating neurons, while in the cytoplasm JNK phosphorylates proteins that slow down migration. Thus the subcellular location of activated JNK is consequential in terms of functional outcome.
4.3 Other Evidence for JNK Regulation of Radial Migration
Interestingly, while Jnk1 −/− mice present with accelerated multipolar and radial migration (Westerlund et al. 2011), Mizuno and colleagues obtained similar results from neuronal progenitor cells in vitro and in developing brain (Mizuno et al. 2005). They found that the vasoactive peptide endothelin-1 induced JNK activity via the Gq heterotrimeric G protein, which activates phospholipase C. They further showed that endothelin-evoked inhibition of radial migration was reversed when the JNK inhibitor JBD was expressed.
4.4 Upstream of JNKs, MAP2Ks Regulate Liver Development and Cortical Migration
Upstream regulators of JNK, MKK4 and MKK7 play essential roles in liver morphogenesis. In contrast to Jnk knockout mice, genetic disruption of Mkk4 or Mkk7 results in defective liver development and poor survival as embryos die between E11.5 and E12.5, displaying defects in hepatocyte proliferation (Table 3.1) (Ganiatsas et al. 1998; Nishina et al. 1999; Yang et al. 1997a; Wada et al. 2004). Strikingly, the Mkk4 −/− and Mkk7 −/− phenotypes match closely that of c-Jun −/− mice (Eferl et al. 1999). However, they diverge noticeably from Jnk1/2 −/− double mutant mice which do not show liver morphogenesis defects, but rather impairment of cranial morphogenesis (Kuan et al. 1999). Early embryonic lethality has prevented more detailed study of the nervous system from Mkk4 −/− mice and targeted deletion strategies have therefore been applied. Targeted disruption of Mkk4 and Mkk7 was accomplished in the nervous system using transgenic Nestin–cre mice. In the case of Mkk4, this resulted in substantial but incomplete loss of MKK4 expression in brain (Wang et al. 2007c). Constitutive JNK activity was reduced postnatally by 80 % and p38 activity by 25 %, indicating a significant impact on downstream signaling events in the postnatal brain. Newborn Mkk4 flox/flox ;Nestin–Cre mice did not exhibit overt differences to their wild-type littermates, however they stopped growing a few days after birth and died within 3 weeks showing defects in growth, balance and righting reflex (Wang et al. 2007c). Substrate phosphorylation was correspondingly reduced in these mice. MAP1B phosphorylation decreased from postnatal day 5 onwards and neurofilament light chain phosphorylation decreased from postnatal day 10 onwards. By adulthood, the mice display axonal tract defects. Specifically, fasciculation of the anterior commissure and corpus callosum is substantially disrupted. While the telencephalon was not studied in great detail, defective positioning of Purkinje cells in the cerebellum was noted.
To further examine MKK7 function in the nervous system, Mkk7 flox/flox ; Nestin–Cre mice were generated with specific deletion of Mkk7 in neural stem cells (Yamasaki et al. 2011). MKK7 expression was significantly reduced from E16.5 onwards, while there was a compensatory gain in MKK4 expression. Mkk7 flox/flox ; Nestin–Cre mice were comparable to wild-type mice up to E18.5 but as with Jip3 −/− mice, they died due to failed respiration (Kelkar et al. 2003). Examination of embryonic brains revealed serious defects in axonal tract formation (reduced size of the corpus callosum, anterior commissures and internal capsule). Electron microscopy examination of axons in Mkk7 flox/flox ; Nestin–Cre mice revealed defects in neurofilaments and the presence of autophagic vacuoles and swollen mitochondria. Consistent with the neurofilament defects, neurons isolated from Mkk7 flox/flox ; Nestin–Cre showed reduced axon length (Yamasaki et al. 2011). In line with this observation, inhibition of JNK in cultured cortical and hippocampal neurons retards axodendritic growth (Tararuk et al. 2006; Oliva et al. 2006) and Jnk1 −/− mice show disrupted axon tracts (Chang et al. 2003). Detailed inspection of the brain tissue revealed enlarged ventricles, reduced striatum, and severe defects in axon formation. The features were not observed in the Mkk4 flox/flox ; Nestin–Cre, thereby implying that MKK4 and MKK7 can exert functionally distinct outcomes. Moreover genetic disruption of Mkk7 in the nervous system moderately altered radial migration.
In conclusion, disruption of either Mkk4 or Mkk7 in the CNS, leads to a reduction in JNK activity in the brains of these mice and reduced phosphorylation of the JNK substrates c-Jun, MAP1B and NFL. However, the temporal influence of these kinases on substrate phosphorylation differs, MKK7 playing a critical phosphorylation role at earlier developmental time points. These studies also show that MKK7 and MKK4 can play differential roles in directing JNK substrate phosphorylation, MKK7 is a critical upstream component in DCX phosphorylation while MKK4 seems to play no role in targeting JNK towards this substrate (Wang et al. 2007c; Yamasaki et al. 2011).
4.5 MEKK1 and DLK Regulate Migration Upstream of MAP2K
Several upstream regulators of JNK signalling were identified, most of which have not been systematically studied in the nervous system. Among those studied however, MEKK4 and DLK stand out because genetic deletion of these genes results in defective cell migration during cortical formation. MEKK1 (MEK kinase 1) is a MAP3K that directly phosphorylates and activates MKK4 and MKK7, though it has a preference for MKK4. MEKK1 therefore activates p38 and JNK but also ERK (Kyriakis and Avruch 2012). MEKK1 is essential for embryonic stem cell migration, keratinocyte migration. Mice lacking MEKK1 display impaired eyelid closure due to disturbed epithelial cell movement, these mice are born with open eyes (Xia et al. 2000; Xia and Kao 2004). JNK activity is reduced by 50 % in Mekk1 −/− mice and JNK is thought to contribute to the eyelid closure phenotype (Takatori et al. 2008). MEKK1 co-localizes with α-actinin along actin stress fibers (Christerson et al. 1999) suggesting an involvement of the actin cytoskeleton in MEKK1-regulated cell movement. On the other hand, MEKK1 activates JNK in cells treated with microtubule toxins and targeted disruption of Mekk1 in embryonic cells results in loss of JNK function and increased apoptosis, suggesting that MEKK1 can protect cells from apoptotic death (Yujiri et al. 2000), possibly as a consequence of disturbed microtubule integrity. While MEKK1 can regulate migration in non-neuronal cells via JNK (Xia and Kao 2004; Xia et al. 2000), there is no evidence that MEKK1 regulates migration in neurons.
MEKK4, like MEKK1, is a MAP3K that can regulate JNK and p38 activity (Takekawa et al. 2005). Mice lacking Mekk4 develop neural tubes defects including cranial exencephaly, spina bifida and curly tail. They also present with severe periventricular heteropia (Chi et al. 2005). The activity of MKK4 was decreased in these embryos, as was p38, however JNK activity remained unchanged. Mekk4 −/− embryos showed massively elevated apoptosis before and during neural tube closure. Rakic and colleagues subsequently showed that siRNA knockdown of Mekk4 using siRNA retarded neuronal migration and BrdU-labelling of Mekk4 −/− embryos at E14.5 reveals impaired exit of BrdU positive cells from the ventricular zone (VZ) (Sarkisian et al. 2006). Importantly however, while p38 was strongly inhibited, the JNK pathway activity was unaltered in Mekk4 −/− brain, and phosphorylation of the JNK substrate DCX was unaffected (Sarkisian et al. 2006; Abell et al. 2005). This data indicate that MEKK4 is required for neuroepithelial cell survival early and radial migration later during cortical development. Impaired migration in Mekk4 −/− embryos correlates with upregulated Filamin A expression and elevated neuroepithelial cell apoptosis may be due to loss of GADD45-induced p38 activation (Takekawa et al. 2005). Thus MEKK4 is a critical regulator of radial migration during corticogenesis. Its mechanism of action most likely involves p38 as JNK activity is not altered in Mekk4 −/− brain (Chi et al. 2005).
DLK is a MAP3K that is highly expressed in developing brain and spinal cord (Hirai et al. 2006). In C. elegans DLK regulates MKK4 and p38 activity and is required for nerve regeneration (Nakata et al. 2005; Hammarlund et al. 2009). In fly and mouse, DLK activation promotes stress responses and Wallerian degeneration (Miller et al. 2009; Ghosh et al. 2011). DLK has also been associated with radial migration regulation in the developing neocortex. At E16, DLK expression is concentrated in the IZ of the telencephalon while by E18, it accumulates in the SVZ (Hirai et al. 2002). Ectopic expression of DLK using adenoviral gene transfer to E13 mice activates JNK and arrests cell migration in the SVZ, while transfer of a kinase dead DLK had no effect. This data implied that DLK negatively regulated radial migration. However, in contrast, it was later reported that radial migration was arrested in Dlk1 −/− mice or upon treatment with SP600125, an ATP-competitive kinase inhibitor of JNK (Hirai et al. 2006). A second study also showed that SP600125 retarded radial migration in cortical slices (Kawauchi et al. 2003). These results were attributed to JNK inhibition, however results obtained using SP600125 are difficult to interpret, as this molecule is not specific for JNK. A kinome screen has shown that SP600125 inhibits 74 kinases (of 353 tested) at a concentration of only 10 μM. Among those kinases inhibited are several MAPK pathway upstream regulators e.g. MEK1, MEK2, MKK3, MKK4 and MKK6 [results from KINOMEscan Library of Integrated Network based Cellular Systems (LINCS) data base]. Nonetheless, these studies highlighted an important role for DLK in regulating radial migration (Hirai et al. 2002; Kawauchi et al. 2003).
5 JNK and Cell Migration – A Closer Look
Several lines of evidence have shown that JNK is required for migration in a variety of cell types including epithelial cells, fibroblasts, endothelial cells, various cancer cell lines and in aortic vascular smooth muscle cells (Huang et al. 2003; Malchinkhuu et al. 2005; Björkblom et al. 2012; Kavurma and Khachigian 2003). This large and concurrent data set derives largely from wound healing assays and the use of Jnk −/− cells, or pharmacological inhibition (Javelaud et al. 2003; Huang et al. 2003). The situation in neurons has been less clear however. Studies of mouse genetics have revealed that components of the JNK cascade namely JNK1, MEKK4 and DLK influence neuronal migration in the cortex, albeit in different directions (inhibitory versus facilitatory). In freshly isolated cerebellar granule neurons and in cortical neurons, JNK1 activity retards migration as demonstrated using Jnk −/− cells or upon expression of the JNK inhibitor JBD (Westerlund et al. 2011; Björkblom et al. 2012). These authors also find that in non-neuronal cells JNK facilitates migration, thus inhibitors of JNK retard motility, in agreement with a large literature. Interestingly, ectopic expression of a neuron-enriched JNK substrate (MARCKSL1) in fibroblasts reverses this migration phenotype to one that mimics JNK action in neuronal cells, where JNK retards migration (Björkblom et al. 2012). Therefore the conflicting reports as to whether JNK activates or inhibits migration in neuronal and non-neuronal cells may depend on JNK target expression. Perhaps the functional multiplicity displayed by JNK in controlling cell migration is not altogether surprising given the signaling diversity at the MAP3K and MAP4K level of the JNK cascade, the components of which least well defined in neurons and brain tissue. The striking differences in phenotypes, while comparing even MAP2K (Mkk4 and Mkk7) single knockouts with MAPK (Jnk) knockout mice, indicates that cooperative activation of JNKs by MKK4 and MKK7 may be critical for JNK’s management of migration in neuronal cells.
5.1 JNK’s Association with Controllers and Executors of Neuronal Migration
DCX: It is generally accepted that MAPK cascades function linearly, the MAPK is the effector kinase and the upstream kinases do not signal orthogonally. The substrate of the effector kinase therefore reveals information on mechanism. Among the well-characterised substrates for JNKs are several microtubule-modifying proteins, including the classical neuronal migration protein doublecortin, also known as DCX (Fig. 3.2). DCX is a developmentally regulated, brain-specific microtubule-associated protein that was first identified in patients with missense mutations in DCX and lissencephaly syndrome (characterized by smooth brain), subventricular heteropia and cortical dysgenesis (des Portes et al. 1998; Francis et al. 1999). DCX protein is phosphorylated by JNK on T331, S334 and T321 (sites from human DCX isoform 2) (Gdalyahu et al. 2004) and phosphorylation of these sites is important for neurite length regulation and neuronal migration (Gdalyahu et al. 2004; Bai et al. 2003). DCX binds to microtubules and enhances polymer formation, it may do this by regulating microtubule nucleation (Fourniol et al. 2010). More recently, DCX has been shown to regulate F-actin through its C-terminal region and this is thought to involve its interaction with spinophillin (the F-actin-binding, regulatory subunit of protein phosphatase 1). Crosslinking of actin and microtubules by DCX is proposed to be important in growth cone guidance as DCX −/y ;Dclk1 −/− mutant neurons are unresponsive to netrin guidance cues (Fu et al. 2013). Furthermore, DCX is required for multipolar transit and bipolar cell locomotion (LoTurco and Bai 2006). DCX phosphorylation by JNK2 may dissociate it from microtubules (Jin et al. 2010) thereby increasing microtubule plasticity and influencing migration. Whether JNK1 or JNK3 isoforms phosphorylate DCX in developing brain and alter migration rate remains to be seen.
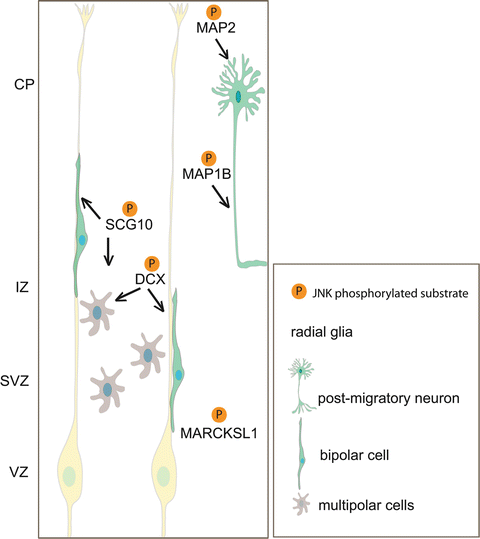
Fig. 3.2
Model depicting neuronal JNK substrates and their respective roles in regulating neuronal migration. JNK phosphorylates SCG10 and DCX, microtubule regulatory proteins that have a major influence on multipolar transition and bipolar cell migration. When neurons have reached their final position, JNK phosphorylation of MAP1b and MAP2 regulates axonal extension and dendritic arbor development. MARCKSL1 is also phosphorylated by JNK and regulates neuronal migration in vitro (Björkblom et al. 2012). Whether MARCKSL1 regulates radial migration remains to be seen. CP cortical plate, IZ intermediate zone, SVZ subventricular zone, VZ ventricular zone
Reelin: Reelin is an extracellular glycoprotein that negatively regulates migration in the developing cortex, hippocampus and cerebellum giving rise to the Reeler mouse phenotype (Tissir and Goffinet 2003). An important link between Reelin and JNK signalling has been established. Mutations in Reelin or in two of its multiple receptors APOER2 and VLDL (Trommsdorff et al. 1999), or in the DAB1 protein that serves as intracellular adaptor protein leads to a lissencephaly in humans (Trommsdorff et al. 1999; Hong et al. 2000). A proline rich region in the cytoplasmic tail of APOER2 binds to two of the JNK scaffold proteins, JIP1 and JIP2. The function of the JIP scaffolds is to co-assemble JNK and distinct MAP2Ks and MAP3Ks thereby facilitating the activation of JNK by these upstream regulators (Manning and Davis 2003; Whitmarsh 2006). Importantly binding of JIP to APOER2 does not disturb the assembly of a complete JNK signalling module consisting of its upstream activators MLK3 and MKK7 (Stockinger et al. 2000). Therefore APOER2 is capable of organizing a scaffold signalling complex that activates JNK, and it could be expected that Reelin binding to its receptor will regulate JNK activation, though this still requires experimental validation. Reelin also controls neuroblast migration to the olfactory bulb via the receptor APOER2 (Hellwig et al. 2012). Whether or not JNK regulates cell motility in the rostral migratory stream is not known.
Notably, Reeler mice mimic Jnk1 −/− mice in several significant ways. Firstly, multipolar and bipolar cells in Reeler mice move faster than in wild-type mice and transit more quickly through the multipolar phase (Britto et al. 2011). This mimics the accelerated migration of multipolar and bipolar cells in Jnk1 −/− mice with reduced duration of the multipolar phase (Westerlund et al. 2011). Another feature of migration in Reeler mice was the disturbed trajectories of migrating neurons that exhibited increased meandering (Britto et al. 2011). Whether neuronal trajectories are altered during cortical development in Jnk1 −/− mice has not been reported. Finally, Reeler mice are characterized by abnormal reorganisation of preplate (PP) neurons, exemplified by delayed and incomplete PP splitting (Sheppard and Pearlman 1997). Similarly, Jnk1 −/− mice display more prominent chondroitin sulphate proteoglycan staining in the superplate that may reflect differences in PP splitting (Westerlund et al. 2011). Together these findings point to JNK as a likely intracellular purveyor of Reelin’s stop signal during formation of the cortex.
5.2 JNK1 Regulation of Microtubules
The microtubule cytoskeleton and its posttranslational modifications are critical determinants of neuronal migration and there are several examples of mutant mice, where disruption of genes that directly or indirectly affect microtubule homeostasis impose migration defects. For example, missense mutations in DCX (des Portes et al. 1998; Francis et al. 1999), TUBA1A (Kumar et al. 2010), TUBG1 (Poirier et al. 2013) and Elongator (Creppe et al. 2009) result in significantly altered migration patterns in the developing cortex. In this context, JNK1 has emerged as a prominent regulator of microtubule integrity in brain. Microtubule length is decreased in brains from adult mice lacking Jnk1 (Chang et al. 2003), microtubule dynamics is altered in cortical neurons upon expression of the JBD inhibitor of JNK (Tararuk et al. 2006), and there is a significant increase in tyrosinated tubulin in Jnk1 −/− neonates, suggesting increased microtubule plasticity (Westerlund et al. 2011). Consistent with this function in maintaining brain microtubule stability, axon formation is also disturbed in Jnk1 −/− mice, for example, the anterior commissure develops normally but is disrupted by postnatal day 12 and is absent in adult brain (Table 3.1) (Chang et al. 2003).
Related posts:
 The Dynamics of Neuronal Migration
Spinal Motor Neuron Migration and the Significance of Topographic Organization in the Nervous System
Mark/Par-1 Marking the Polarity of Migrating Neurons
The PAR Polarity Complex and Cerebellar Granule Neuron Migration
Molecular Pathways Controlling the Sequential Steps of Cortical Projection Neuron Migration
Extracellular Signals Controlling Neuroblast Migration in the Postnatal Brain
The Dynamics of Neuronal Migration
Spinal Motor Neuron Migration and the Significance of Topographic Organization in the Nervous System
Mark/Par-1 Marking the Polarity of Migrating Neurons
The PAR Polarity Complex and Cerebellar Granule Neuron Migration
Molecular Pathways Controlling the Sequential Steps of Cortical Projection Neuron Migration
Extracellular Signals Controlling Neuroblast Migration in the Postnatal Brain
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


