We will consider separately the pathobiology associated with mtDNA mutations and with nDNA mutations.
PATHOBIOLOGY OF mtDNA MUTATIONS: MITOCHONDRIAL GENETICS VERSUS MENDELIAN INHERITANCE
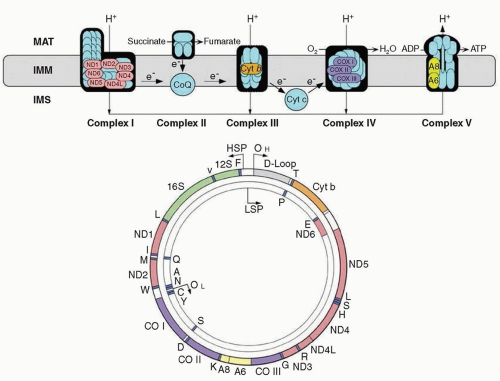
Human mtDNA is a small (16.6 kb) circle of double-stranded DNA (
Fig. 139.1) comprising only 37 genes. Of these, 13 encode polypeptides, all subunits of the respiratory chain: 7 subunits of complex I (NADH-ubiquinone oxidoreductase), 1 subunit of complex III (ubiquinone-cytochrome
c oxidoreductase), 3 subunits of complex IV (cytochrome
c oxidase [COX]), and 2 subunits of complex V (ATP synthase). The other 24 genes encode 22 transfer RNAs (tRNAs) and 2 ribosomal RNAs (rRNAs) that are required for translation of messenger RNAs on mitochondrial ribosomes
(mitoribosomes). The subunits of complex II (succinate dehydrogenase [SDH]-ubiquinone oxidoreductase) and the enzymes required for synthesis of small electron carriers (coenzyme Q
10 [ubiquinone] and cytochrome
c) are encoded exclusively by nDNA.
The following main principles distinguish mitochondrial genetics from mendelian genetics and help explain many of the clinical peculiarities of mtDNA-related disorders.
Polyplasmy. Most cells contain multiple mitochondria and each mitochondrion contains multiple copies of mtDNA so that there are thousands of mitochondrial genomes in each cell.
Heteroplasmy. When an mtDNA pathogenic mutation affects some but not all genomes, a cell, a tissue, indeed a whole individual will harbor two populations of mtDNA, normal (or wild-type) and mutant, a condition known as heteroplasmy; in normal tissues, all mtDNA were considered identical, a situation called homoplasmy. However, highly sensitive next-generation sequencing has revealed the coexistence of mutated mtDNA variants (0.2% to 2.0% heteroplasmy) and wild-type mtDNA within a cell in blood and skeletal muscle of clinically unaffected individuals (a phenomenon termed universal heteroplasmy).
Threshold effect. The functional impairment associated with a pathogenic mtDNA mutation is largely determined by the degree of heteroplasmy, and a minimal critical number of mutant genomes (the mutation load) is required before tissue dysfunction becomes evident (and related clinical signs become manifest), a concept aptly termed threshold effect. This is, however, a relative concept: Tissues with high metabolic demands, such as brain, heart, and muscle, tend to have lower tolerance for mtDNA mutations than do metabolically less active tissues.
Mitotic segregation. Both organellar division and mtDNA replication are apparently stochastic events unrelated to cell division: Thus, the number of mitochondria (and mtDNA) can vary not only in space (i.e., among cells and tissues) but also in time (i.e., during development or aging). Moreover, at cell division, the proportion of mutant mtDNAs in daughter cells may drift, allowing for relatively rapid changes in genotype; therefore, if and when the threshold is exceeded, a clinical phenotype may manifest.
Maternal inheritance. At fertilization, all mitochondria (and all mtDNA) in the zygote are derived from the oocyte. Therefore, a mother carrying an mtDNA pathogenic mutation will pass it on to all her children, males and females, but only her daughters will transmit to their progeny, in a vertical matrilineal line. When maternal inheritance is evident in a clinical setting, it provides strong evidence that an mtDNA mutation must underlie the disease in question. However, the other features of mitochondrial genetics (e.g., heteroplasmy degree and
the threshold effect) often mask maternal inheritance by causing striking intrafamilial clinical heterogeneity. Thus, when suspecting an mtDNA-related disorder, it is crucial to collect the family history meticulously, paying special attention to “soft signs” (such as short stature, hearing loss, or migraine headache) in potentially oligosymptomatic maternal relatives.
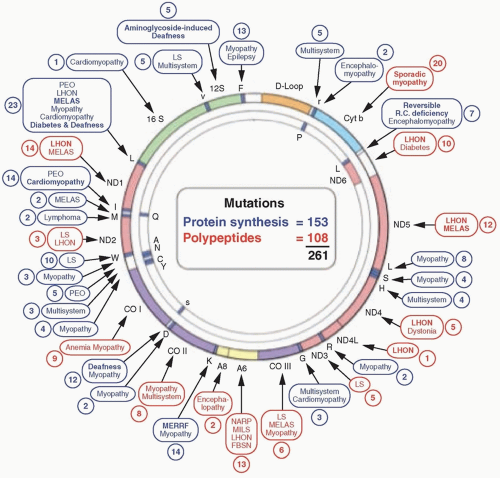
More than 250 point mutations of mtDNA have been identified (
Fig. 139.2). Ten pathogenic mutations in mtDNA have been observed frequently in neonatal cord blood of over 3,000 well babies, and a surprisingly high frequency, 1 in 200, harbored a pathogenic mtDNA mutation. The frequent occurrence of mtDNA mutations leads to a correspondingly high prevalence of mtDNA-related diseases: In the northeast of England, for example, 1 in 10,000 people are clinically affected by mtDNA-related disorders, and 1 in 6,000 individuals are considered at risk.
MENDELIAN INHERITANCE
The identification of nDNA mutations for mitochondrial diseases has accelerated rapidly since the discovery in 1995 of the first “direct hit” mutation in an nDNA-encoded gene for one of four subunits of complex II, exclusively controlled by nuclear genetics. These advances have been made possible by rapidly evolving technologies, from linkage analysis, homozygosity mapping, sequencing of candidate genes, and—ultimately—to next-generation genome-wide (whole-exome) sequencing (WES) or to screening of the portion of the genome that encodes the approximately 1,700 mitochondrial
proteins in a process known as
mito-exome sequencing. The advantages of high-throughput DNA sequencing are eminently practical, enabling rapid diagnosis of puzzling cases and informing genetic counseling.
Direct Hits Mutations
“Direct hits” refer to pathogenic mutations that directly affect nDNA-encoded respiratory chain subunits in all five complexes but most commonly affecting the 16 of the 45 structural subunits of the gigantic complex I. In contrast, only a few direct hits affect complex II (affecting any of the four subunits), complex III (2 of the 11 subunits harbor damaging mutations, UQCRB and UQCRQ), complex IV (COX), of which only COX subunits 6B1 and 7B harbor deleterious mutations, and complex V (ATP synthase, containing a direct hit in a single gene, ATP5E). In keeping with the all-or-none effect of mostly recessive mendelian mutations, as opposed to the variegated effect of heteroplasmic mtDNA mutations, disorders to direct hits to the mitochondrial respiratory chain generally manifest at or soon after birth and are severe, often being lethal in infancy, most often associated with Leigh syndrome.
Indirect Hits Mutations
Even when all nDNA-encoded subunits of the various complexes are expressed correctly, they must be translated, imported into mitochondria, and directed to the inner mitochondrial membrane (IMM). At this site, they assemble with their mtDNA-encoded counterparts, acquire prosthetic groups, if necessary multimerize, and further assemble into supercomplexes (respirasomes). Mutations in these pathways are referred to as indirect hits, as they indirectly affect the mitochondrial respiratory chain.
In 1998, the search for the molecular basis of COX-deficient Leigh syndrome led to simultaneous discovery by two groups of the first mutant mitochondrial assembly gene, SURF1. Mutations in SURF1 are among the most common causes of Leigh syndrome. Mutations in at least seven more COX assembly factors (SCO1, SCO2, COX10, COX14, COX15, COA5, FAM36A, and TACO1) have been associated with various human diseases, predominantly encephalopathy, but cardiopathy often occurs with some mutations (SCO2, COX10, COX15, and COA5), whereas hepatopathy is associated with the SCO1 mutations.
In patients with complex I deficiencies that are difficult to define molecularly, mutant assembly factors have been identified through next-generation sequencing of DNA. The clinical manifestations of these indirect hits tend to be more clinically heterogeneous than those associated with direct hits; all described patients have had encephalopathy resembling Leigh syndrome, but many have had leukodystrophy rather than gray matter involvement. Cardiomyopathy is often the dominating feature, presenting in infancy or in early childhood and early death.
The first assembly defect in complex III was identified in 2002 in Finnish infants with an extremely severe syndrome named GRACILE, an acronym aptly summarizing the symptoms and signs: growth retardation, aminoaciduria, cholestasis, iron overload, and early death. The mutated protein was the mitochondrial chaperone BCSL1, needed for insertion of the iron-sulfur (Fe-S) subunit into complex III.
A new group of disorders attributed to deficiency of coenzyme Q10 (CoQ10) can be related to indirect hits that consist of mutations in a cascade of biosynthetic enzymes. They result in deficiency of this relatively simple component, CoQ10, which is an integral part of the respirosome, where it shuttles electrons from complex I and complex II to complex III, acts as an antioxidant and modulates apoptosis. Molecular defects in genes encoding CoQ10 biosynthetic enzymes (PDSS2 and COQ2) were initially discovered in 2006, but mutations in other biosynthetic genes (PDSS1, COQ6, ADCK3, and COQ9) followed. Five clinical syndromes are attributed to CoQ10 deficiency and they will be considered in the following section.
Defects of mtRNA Translation
The mitochondrial genome is transcribed into 13 mRNAs, which are translated by mitoribosomes into the 13 mtDNA-encoded respiratory chain subunits. As described in a comprehensive review, this process can be divided into two phases: a posttranscriptional phase (involving tRNA modifications, aminoacyl-tRNA synthetases, and processing of ribosomal proteins) and mtRNA translation (initiation, elongation, termination, and recycling). Representative examples of defects of each step of the mitochondrial translation process are briefly highlighted.
ABNORMAL tRNA MODIFICATIONS
Pseudouridylation modification is thought to promote stability and conformation of mitochondrial and nuclear tRNAs. Mutations in pseudouridine synthase 1 (PUS1) differentially impair nuclear and mitochondrial tRNA maturation causing mitochondrial myopathy, lactic acidosis, and sideroblastic anemia (MLASA); in addition, psychiatric symptoms and facial dysmorphism are likely due to abnormal cytosolic protein synthesis.
The conundrum of spontaneous improvement in infantile reversible COX-deficient myopathy (more accurately designated reversible infantile respiratory chain deficiency [RIRCD]) was resolved by elucidation of the tRNA modification processes in this disorder. RIRCD is due to homoplasmic mutations in MT-TE, which encodes mt-tRNAGlu. Pathogenesis of the disease became more complex when patients in two families were found to harbor mutations in the TRMU gene, which modifies a wobble uridine base in mt-tRNAGlu. Skeletal muscle from infants in the initial symptomatic phase showed significantly decreased 2-thiouridylation due to defective TRMU activity, which exacerbated the effect of the mtRNAGlu mutation and triggered the mitochondrial translation defect, thus demonstrating an nDNA-modifying effect on the mtDNA mutation.
Mutations in nine highly specialized aminoacyl-tRNA synthetases (ARs), which covalently link an amino acid to its cognate tRNA (tRNA charging), are associated with multiple specific clinical syndromes. Mitoribosomes consist of two mRNA components (12S and 16S rRNAS) and of novel about 30 and 50 nDNA-encoded proteins. Of the roughly 80 ribosomal proteins, 3 have been firmly linked to mitochondrial diseases: MRPS16, MRPS22, and MRPL3. Patients with defects of mitochondrial elongation factor genes (
GFM1 encoding EFG1;
TUFM, EF-Tu
mt; and
TSFM, EF-TS
mt) typically present with infantile-onset multisystemic diseases. Of the three known mitochondrial translation termination factors, which catalyze release of fully synthesized polypeptides, mutations have only been identified in a poorly characterized ribosome-recycling factor encoded by
C12orf65 in two unrelated families with LS, optic atrophy, and ophthalmoplegia. Mutations in two mitochondrial translation activators, both required for COX, have been linked to human diseases. Mutations in the gene
TACO1 (encoding translational activator for COX I), cause late-onset Leigh syndrome and COX deficiency, whereas mutations in
LRPPRC (which encodes a leucine-rich pentatricopeptide repeat-containing protein) have been identified as the cause of a distinctive Leigh syndrome prevalent in French Canadians. Mutations of
RMND1 cause mitochondrial translational defects affecting multiple organs; however,
the function of the encoded protein, which contains a domain of unknown function (DUF 155), remains a mystery.
Defects of the Inner Mitochondrial Membrane Lipid Milieu
The respiratory chain resides in the phospholipid component of the IMM and alterations in the lipid milieu are increasingly associated with mitochondrial encephalomyopathy. Cardiolipin, a dimeric molecule composed of two phosphatidylglycerol moieties connected by a glycerol group, is the signature mitochondrial phospholipid and a major component of the IMM, where it is synthesized. Cardiolipin deficiency was first documented—together with mutations in the gene encoding tafazzin (TAZ)—in patients with Barth syndrome, an X-linked mitochondrial myopathy and cardiomyopathy, with neutropenia and stunted growth.
Similar to Barth syndrome, Sengers syndrome affects primarily heart and muscle, with the distinctive additional clinical feature of congenital cataracts. Whole exome sequencing revealed mutations in the acylglycerol kinase (AGK) gene. Notably, lack of phosphatidic acid is a precursor of cardiolipin, thus providing a point of convergence in Sengers syndrome and Barth syndrome, which could account for the clinical similarities between the two disorders.
The mitochondrial-associated endoplasmic reticulum (ER) membrane (MAM) is the close physical and functional association between ER and mitochondria. MAMs are involved in multiple functions, including lipid transport, cholesterol metabolism, calcium signaling, energy metabolism, apoptosis, and mitochondrial dynamics.
New diseases have been associated with MAM dysfunction, including the MEGDEL syndrome (3-methylglutaconic aciduria type IV, deafness, and Leigh syndrome), in which the SERAC protein is located in the MAM and controls exchange of phospholipids between the ER and mitochondria. Interestingly, a central role of MAM in the pathogenesis of Alzheimer disease has been well documented.
Defects of Mitochondrial Dynamics
Mitochondria are highly dynamic organelles that move around the cell on microtubular tracks and, similar to their bacterial precursors, they fuse and divide, thus forming tubular networks that favor a balanced distribution of energy throughout the cell. Impairment of mitochondrial motility, fusion, or fission causes the nervous system particularly susceptible, as it is highly dependent on oxidative energy, and mitochondria must travel long distances along central axons and peripheral nerves.
Mitochondrial fusion requires coordinated action of multiple guanosine triphosphatases (GTPases) in the outer mitochondrial membrane (OMM), namely mitofusins MFN1 and MFN2, and in the IMM, namely OPA1.
Mitochondrial fission requires cytosolic dynamin-related protein 1 (DRP1; also a GTPase), which is recruited to the OMM together with two partners, mitochondrial fission (FIS1) and mitochondrial fission factor (MFF). Once recruited to the OMM, DRP1 forms a spiral that wraps around the mitochondrion like a noose and severs the mitochondrial membrane using guanosine triphosphate (GTP) hydrolysis.
DEFECTS OF MITOCHONDRIAL MAINTENANCE
These disorders have been traditionally subdivided into two groups: (1) mtDNA depletion syndromes manifesting early in life and presenting as myopathy, hepatocerebral disease, or encephalomyopathy and (2) multiple mtDNA deletions, usually associated with chronic progressive external ophthalmoplegia with other manifestations that are predominantly neurologic (progressive external ophthalmoplegia [PEO]-plus).
The disorders of intergenomic communication are usually caused either by defects of mtDNA replication that are catalyzed by polymerase γ (genes POLG and POLG2) and by the helicase Twinkle (PEO1) or by altered homeostasis of the intramitochondrial deoxynucleoside triphosphates (dNTPs) pool that is controlled by multiple genes including TK2 (encoding thymidine kinase 2), DGUOK (encoding deoxyguanosine kinase), and TYMP (encoding thymidine phosphorylase). In addition, the gene encoding adenine nucleotide translocase 1 (ANT1) causes autosomal dominant progressive external ophthalmoplegia (adPEO). Another mutant gene (OPA1), which is required mitochondrial fusion, has curiously been associated with autosomal dominant PEO-plus.
It is evident that mtDNA depletion and multiple mtDNA deletions often coexist in the same patient and are often caused by the same genes that cause either isolated depletion or multiple deletions of mtDNA (
Table 139.2). A peculiarity of these disorders is that—although they are unequivocally mendelian—they share many features of mitochondrial genetics, including heteroplasmy and the threshold effect.