Mitochondrial Encephalomyopathies
Harvey B. Sarnat
John H. Menkes
Mitochondrial disease may cause any symptom in any tissue at any age by any inheritance.
—A. Munnich
The above definition of mitochondrial diseases summarizes the clinician’s dilemma in diagnosing this most ubiquitous of metabolic disorders, with pathogenic mitochondrial DNA mutations being found in at least 1 in 8,000 individuals (1). Nevertheless, there are some patterns of specific mitochondrial syndromes that can be recognized at least enough to justify the extensive laboratory investigations required for confirmation. Because mitochondria are essential organelles of nearly all cells, with the exception of mature erythrocytes, mitochondrial diseases may affect any organ system and for this reason are now termed mitochondrial cytopathies. The most frequently affected organs are striated muscle, central nervous system, and heart. Diagnostic changes may be demonstrated in striated muscle even in patients who do not overtly manifest myopathic symptoms and signs; hence the muscle biopsy can be valuable diagnostic procedure.
ORIGIN OF MITOCHONDRIA
Mitochondria were free-living independent organisms with their own unique DNA and membranes about 2 billion years ago, when they developed a symbiotic relationship with the evolving proeukaryocytes, and the two have lived more or less harmoniously as eukaryotic cells ever since. The cell depends on its mitochondria for the generation of oxidative enzymes of aerobic energy production, and the mitochondria depend upon the cell’s nuclear DNA to provide the majority of the subunits for its respiratory chain complexes.
HISTORY OF DISCOVERY OF MITOCHONDRIA AND DISEASES OF THE ORGANELLE
The first probable human visualization of mitochondria was in 1841, when intracellular granules were described using the crude optics of early microscopes. In 1890 Altmann (2) recognized the ubiquitous nature of “bioblasts” as elementary organs living in cells, and in 1898 Benda named the organelle the “mitochondrion” (3). The ultrastructure of mitochondria was described in 1952 by Palade (4), and mitochondrial DNA (mtDNA) was defined by Nass and Nass in 1963 (5). Luft was the first to describe a mitochondrial disease in 1962, deciphering a deficit in oxidative phosphorylation in a patient with a now rare disease (6). Ragged-red fibers were described in the muscle biopsy and recognized as associated with mitochondrial disorders in 1963 by Engel and Cunningham (7). In the early 1970s, several disorders of brain and muscle involving deficiencies in respiratory chain enzymes were described, and in 1973, DiMauro and DiMauro provided the first examples of myopathies due to isolated deficiencies of muscle carnitine and carnitine palmitoyltransferase (8). By 1981, the complete genomic sequence of human mtDNA was documented (9). In 1984 phenotype–genotype correlations were being made by Pavlakis et al. (10), and the concept of mitochondrial syndromes was founded. In 1988, major advances occurred with the description by Holt et al. (11) of human encephalomyopathies associated with large deletions of mtDNA and the description by Wallace et al. (12) of Leber hereditary optic neuritis as due to a point mutation at nucleoprotein (np)11778 in mtDNA, a clinical disorder subsequently shown to be a syndrome associated with several additional mtDNA point mutations in other patients, as is the case with most mitochondrial disorders. Hundreds of point mutations or deletions of mtDNA have been described since 1990, and new genetic varieties are continuously being discovered. Mitochondrial diseases also can result from mutations in the nDNA that encodes respiratory chain subunits or parts of mitochondrial membranes,
even with a normal mtDNA genome. In 2003, Servidei showed that large-scale mtDNA rearrangements, rather than mtDNA deletions or point mutations, are the basis of some mitochondrial diseases (13). Nearly all of the distinctive clinical and pathologic patterns of mitochondrial diseases were initially thought to be singular diseases but now are recognized as syndromes in which the same phenotype can be produced by multiple mitochondrial genotypes.
even with a normal mtDNA genome. In 2003, Servidei showed that large-scale mtDNA rearrangements, rather than mtDNA deletions or point mutations, are the basis of some mitochondrial diseases (13). Nearly all of the distinctive clinical and pathologic patterns of mitochondrial diseases were initially thought to be singular diseases but now are recognized as syndromes in which the same phenotype can be produced by multiple mitochondrial genotypes.
FUNCTIONS OF MITOCHONDRIA
The biochemical aspects of mitochondrial structure and function are reviewed more extensively by Lodish et al. (14) and Schapira (15). The primary function of mitochondria is to generate the high-energy phosphate bond in ATP by phosphorylation of adenosine diphosphate (ADP). The energy required for this reaction is derived from oxidation of the metabolic products of carbohydrates, fatty acids, and proteins. These processes of energy generation are collectively referred to as oxidative phosphorylation. Energy production is the only function of mitochondrial DNA; unlike the nuclear DNA of the cell, mtDNA does not determine any features of anatomic structure of the body, growth, or tissue differentiation.
Mitochondrial oxidative phosphorylation involves electron transport among five respiratory chains of enzymes, termed respiratory complexes. Complex I is nicotinamide adenine dinucleotide diaphorase; complex II is succinate dehydrogenase; complex III is ubiquinone or cytochrome b oxidase; complex IV is cytochrome c oxidase; complex V is ATP synthase. Coenzyme Q10 helps to mediate transport from complex I to complexes III and IV.
Pyruvate dehydrogenase is one of the most complex enzymes known, with a molecular weight of greater than 7 million. It contains multiple subunits of three catalytic enzymes (E1, E2, and E3), two regulatory polypeptides, and five different coenzymes. The pyruvate dehydrogenase complex mediates the oxidation of pyruvate to acetyl coenzyme A to carbon dioxide, to generate reduced electron carriers through a series of nine reactions, carried out within the mitochondrial matrix by the citric acid cycle of Krebs. These reactions are carried out on the inner mitochondrial membrane by the electron transport chain, a set of electron carriers grouped into five multienzyme complexes. The free energy released during oxidation is stored in an electrochemical proton gradient across the inner mitochondrial membrane. The movement of protons back across the mitochondrial membrane is then coupled with the synthesis of ATP from ADP and phosphate. Coenzyme Q10 subserves the transport of electrons from complex I to complexes III and IV.
Transport of free fatty acids into mitochondria as fuel is mediated by carnitine. Within the mitochondria, the fatty acids are uncoupled from carnitine, a carrier molecule, and react with coenzyme A (CoA) to form an acyl-CoA. By a series of four sequential reactions (beta-oxidation), each molecule of acyl-CoA is oxidized to form one molecule of acetyl-CoA and an acyl-CoA shortened by two carbon atoms. Because most defects of beta-oxidation present with organicaciduria, these conditions are considered in Chapter 1 in the section dealing with organic acidurias. Amino acids are oxidized within the mitochondria; after transamination to their respective ketoacids, the amino acids alanine, aspartic acid, and glutamic acid enter the citric acid cycle.
In addition to its principal function in energy metabolism, mtDNA has additional functions during development. It is important for neuroblast polarity by modulating calcium homeostasis in microtubules and also regulates the bcl2 gene for apoptosis. Damage to mtDNA and loss of the mitochondrial membrane potential are demonstrated in apoptotic cell death (16).
GENETICS OF MITOCHONDRIAL STRUCTURE AND DISEASES
The genetics of mitochondrial cytopathies is complex. The mitochondrion has its own DNA (mtDNA) on a single, circular chromosome of about 16.5 kilobases (kb). The mtDNA has an intimate relation with the cell’s nuclear DNA (nDNA), and for each of the five respiratory complexes, the majority of the subunits are encoded by nDNA, not mtDNA. Complex I consists of 41 subunits, of which only 7 are encoded in the mtDNA; the other 34 are encoded in the nDNA. Complex II has only 4 subunits, all of which are encoded in nDNA. Complex III has 10 subunits, 1 encoded by mtDNA and 9 by nDNA. Complex IV has 13 subunits, 3 encoded by mtDNA and 10 by nDNA. Complex V has 12 subunits, 2 encoded by mtDNA and 10 by nDNA. The nDNA encodes the vast majority of the mitochondrial proteins, including all proteins present in the outer membrane and the matrix. The respiratory complexes are located on the inner membrane.
Because in the formation of the zygote, almost all mitochondria are contributed by the ovum, mtDNA is maternally inherited (17). Maternal inheritance appears to be operative in a small but significant proportion of inherited mitochondrial diseases; in the remainder transmission is as an autosomal recessive trait or is sporadic or unpredictable (18). Because the nuclear genome contributes subunits to the respiratory complexes, some mitochondrial diseases may follow a Mendelian pattern of inheritance, in which the metabolic defect is due to defective subunits encoded by mutation in nDNA, with preservation of normal mtDNA. Inheritance in these mutations is nearly always autosomal recessive. Some cases of Leigh encephalopathy provide a good example of this phenomenon. Nine
different genetic defects have been documented in this syndrome, five of which involve mtDNA and four of which involve nDNA. If a point mutation in mtDNA is involved, the obligatory transmission is maternal, though not involving the X chromosome of nDNA.
different genetic defects have been documented in this syndrome, five of which involve mtDNA and four of which involve nDNA. If a point mutation in mtDNA is involved, the obligatory transmission is maternal, though not involving the X chromosome of nDNA.
Homoplasmy refers to the situation in which all mtDNA molecules within the cell are identical, the normal condition. If mutations in mtDNA occur, two populations of mtDNA then coexist, the heteroplasmic condition. Most pathogenic mtDNA mutations are heteroplasmic. In this situation the ratio of normal to abnormal mtDNA determines the clinical expression as well as the severity of the mitochondrial disease. A 100% abnormal mtDNA is incompatible with life. Differences in this ratio of heteroplasmy among cells in different organs determine the clinical expression in each organ system. Heteroplasmy may occur not only at the level of the cell, but also at the level of the individual mitochondrion, intramitochondrial heteroplasmy.
At the time of cell division, mitochondria and mtDNA are randomly partitioned into the two daughter cells, mitotic segregation. The proportion of normal and abnormal mtDNA inherited by each daughter cell is not necessarily equal, and a threshold effect of mtDNA mutation may influence clinical expression including age of onset of symptoms (19).
As an example of heteroplasmy, in the nucleotide (nt)-8993 T→C mutation that substitutes leucine for arginine, if less than 70% of the mtDNA shows this point mutation, the patient has the NARP (nystagmus, ataxia, and retinitis pigmentosa) syndrome; if the mutation involves 90% or more, the patient presents with Leigh encephalopathy. Between 70% and 90% mutated mtDNA produces mixed or variable clinical expression.
Another class of mtDNA abnormalities is large-scale mtDNA rearrangements, such as kilobase deletions of the mitochondrial chromosome. Patients may harbor a 5- or 7.5-kb mtDNA deletion (the most common varieties), with striking differences in the proportion of deleted mtDNA molecules within the total mtDNA population. Sometimes as little as 2% mitochondria with significant deletions may be enough to render the individual symptomatic. High levels of these large-scale mtDNA deletions frequently occur in patients with Kearns-Sayre and PEO (progressive external ophthalmoplegia) syndromes (20). Approximately one-third of Kearns-Sayre patients harbor the same kind of mtDNA deletion, often designated the common 5-kb deletion, but occasional patients with Kearns-Sayre syndrome have larger, 11-kb deletions, with correspondingly more severe neurologic, muscular, and cardiac manifestations (21,22).
Many of the common point mutations are now known, particularly in mitochondrial myopathies with ragged-red fibers, in mitochondrial cardiomyopathies, and in some mitochondrial encephalopathies, especially Leigh encephalopathy, but new mutations are published weekly in this rapidly expanding field of genetics. A table summarizing the known mtDNA point mutations and deletions in mitochondrial encephalomyopathies is updated quarterly in the journal Neuromuscular Disorders (23). Many mitochondrial point mutations are expressed in both striated muscle and central nervous system (CNS). For example, Kearns-Sayre syndrome is clinically mainly a myopathy but also involves the visual system of the brain, demonstrated during life by abnormally slow visual-evoked potentials.
Most genetic laboratories that study mtDNA employ batteries of several point mutations that they screen for the common, well-documented mutations, but cannot test the entire mitochondrial genome. At times, point mutations are found in nucleotide sequences that are not evolutionarily conserved, do not specify highly conserved amino acid residues, and/or are not associated with an amino acid substitution. The interpretation of such defects or polymorphic variants, in the context of clinical and pathologic presentation, often is problematic and uncertain.
CLINICAL SYNDROMES OF MITOCHONDRIAL CYTOPATHIES
Because mitochondria are present in almost every cell, the wide variety of clinical presentations is not surprising. Mitochondrial disorders can be initially classified by clinical criteria in accordance with the organ systems most affected. One principle of mitochondrial diseases in general is that rarely is only one organ affected, and the clinical involvement of multiple organ systems is an important feature that allows the clinician to suspect a mitochondrial disease. The combination of symptoms and signs referable to the central and/or peripheral nervous system and striated muscle is the most frequent. Clinical manifestations are extremely variable, even with a known pathogenic point mutation and even among affected members of the same family. Phenotype–genotype correlations often are poor for the clinical identification of specific mtDNA point mutations or at times for specific mitochondrial syndromes (24,25,26). Multiple genotypes of both mtDNA and nDNA defects may produce similar clinical and pathologic presentations, hence the common patterns of mitochondrial disorders and syndromes.
Each of the mitochondrial syndromes has some constant clinical features that raise suspicion of the diagnosis and justify investigations to confirm or refute this provisional diagnosis. For instance, some patients fit a classical clinical and pathologic phenotype for MELAS (mitochondrial encephalopathy with lactic acidosis and strokelike episodes) but have an mtDNA point mutation that is more
typical for MERRF (myoclonic epilepsy with ragged-red fibers), or they show a mixed MELAS/MERRF phenotype (27,28) or MERRF/Kearns-Sayre phenotype (29). Many mitochondrial cytopathies cannot be classified under a single clinical eponym.
typical for MERRF (myoclonic epilepsy with ragged-red fibers), or they show a mixed MELAS/MERRF phenotype (27,28) or MERRF/Kearns-Sayre phenotype (29). Many mitochondrial cytopathies cannot be classified under a single clinical eponym.
Because each of the mitochondrial syndromes may be a similar clinical expression of multiple genetic defects, and the ratio of normal to abnormal mitochondria is variable between different patients even in the same family and between different organs, the phenotype–genotype correlations often are poor. Myopathology–genotype correlations are somewhat more reliable, but even the muscle biopsy does not always predict the precise genetic mutation.
The neurologic manifestations of mitochondrial diseases are many, but the most common in infancy are seizures, including infantile spasms and myoclonic epilepsy, visual impairment, deafness, central dysphagia, apnea and respiratory failure, diffuse muscular hypotonia, and global developmental delay. In older children and young adults, neurologic deficits include dystonia, cerebellar deficits, spastic diplegia, memory loss, cognitive deficits, progressive hearing loss, dementia, and autism. Regression and loss of skills may be seen as with any neurodegenerative disease. Peripheral neuropathy also occurs in mitochondrial disease and may contribute to the hypotonia and weakness. Because extraocular muscles have more mitochondria per unit volume of tissue than do most other muscles, they often are involved and produce progressive external ophthalmoplegia of all six muscles. Respiratory insufficiency may result from myopathic involvement or from central respiratory failure or a combination.
Lactic acidosis is a prominent feature of many mitochondrial diseases but not all. It is universally present in MELAS syndrome and helps to define this entity but is not present in MERRF and Kearns-Sayre syndromes. It is present in most cases of Leigh encephalopathy but not all; lactic acidosis is more likely in those cases with mtDNA mutations than in cases with nDNA defects. In some patients, the serum lactate may be normal but cerebrospinal fluid (CSF) lactate is elevated. When drawing serum lactate levels, it is important to also measure pyruvate. The most common cause of high serum lactate is that the tourniquet has been left on the arm too long while searching for a good vein; the resulting acute anaerobic state causes pyruvate to become converted to lactate; hence the lactate is high but the pyruvate is very low. In mitochondrial cytopathies, by contrast, if the lactate is elevated, the pyruvate is normal or also high (30). Simultaneous measurement of serum pyruvate will thus remove all doubt about whether an elevated lactate is merely a biological artifact. Other differential diagnoses of high serum lactate are shown in Table 2.1.
Imaging of the brain is an important diagnostic study in patients with mitochondrial cytopathy involving encephalopathy. Characteristic lesions may be seen with both computed tomography (CT) and magnetic resonance imaging (MRI) in the regions of the basal ganglia in particular (Fig. 2.1).
TABLE 2.1 Conditions Producing Lactic Acidosis in Infancy and Childhood | ||||||
|---|---|---|---|---|---|---|
|
Mitochondrial syndromes can be broadly divided into two large categories: those in which the muscle biopsy exhibits ragged-red fibers and those that show many other features of mitochondrial myopathy but not ragged-red fibers. Ragged-red fibers are muscle fibers seen in frozen section and stained with modified Gomori trichrome stain that show irregularly shaped, bright red deposits in the subsarcolemmal and intermyofibrillar sarcoplasm, contrasting with the green myofibrils. The red color with this histochemical stain is derived from the strongly lipophilic chromotrope-2R in the stain, which has a particularly strong affinity for phospholipids; mitochondrial membranes contain a large amount of the phospholipid sphingomyelin. Electron microscopic examination of the ragged-red zone reveals large numbers of closely packed normal and abnormal mitochondria.
A majority of adults presenting clinically with mitochondrial cytopathy have a ragged-red fiber syndrome with combined complex I and IV deficiencies, whereas most infants and children symptomatic with mitochondrial
disease do not have this combination, hence the designation non–ragged-red fiber syndromes. A common combination defect in early childhood is one of complexes III and V, which is unusual in adults (31). Infants with complex I and IV defects may not show ragged-red fibers histologically by trichrome stain until 3 years of age or older; hence this marker is not entirely reliable in infant muscle biopsies.
disease do not have this combination, hence the designation non–ragged-red fiber syndromes. A common combination defect in early childhood is one of complexes III and V, which is unusual in adults (31). Infants with complex I and IV defects may not show ragged-red fibers histologically by trichrome stain until 3 years of age or older; hence this marker is not entirely reliable in infant muscle biopsies.
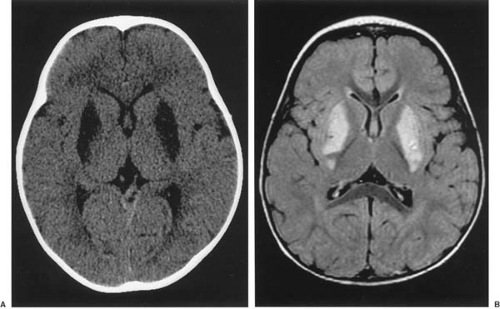 FIGURE 2.1. Imaging of a 21-month-old girl with Leigh encephalopathy. A: Computed axial tomography shows symmetric hypodense areas in the region of basal ganglia. B: Magnetic resonance imaging-T2 FLAIR (fluid attenuation inversion recovery) reveals high signal intensity in the same region. This is a characteristic finding in many mitochondrial diseases with many types of mutations in mtDNA. The muscle biopsy of this child showed no ragged-red fibers but many fibers deficient in cytochrome c oxidase and subsarcolemmal crescents of intense oxidative activity. She had quantitative defects in respiratory chains I, III, and V, but known mtDNA mutations could not be demonstrated. |
RAGGED-RED FIBER MITOCHONDRIAL SYNDROMES
Melas (Mitochondrial Encephalopathy with Lactic Acidosis and Strokelike Episodes)
First described by Pavlakis et al. (10), the clinical onset of this disorder usually is in late childhood or early adult life, but it can present even in infancy. In more than 60% of patients symptoms appear before 15 years of age (32,33). Neurologic deficits are transient ischemic episodes and permanent cerebral infarcts and are referable to vessels as large as the middle cerebral artery or as small as arterioles producing multiple microinfarcts in the brain. The cerebral cortex and subcortical white matter is most frequently involved, particularly in the parieto-occipital region, but the thalamus, corpus striatum, globus pallidus, brainstem, and cerebellum also may be affected with similar lesions. Though all patients with MELAS have strokes, they are not always the initial symptoms. Migrainous headaches, vomiting, seizures, deafness, and focal neurologic deficits are frequent presentations, and some patients suffer progressive dementia (34). The occipital lesions may lead to hemianopia or to cortical blindness. Though muscle is involved pathologically with ragged-red fibers, weakness, myalgias, and other neuromuscular symptoms and signs rarely are evident. Retinitis pigmentosa and cardiomyopathy occur rarely.
The reason for the strokes is the high mutation load of the mtDNA in endothelial cells of cerebral vessels, which leads to deficient pinocytotic vesicles, endothelial swelling, and vascular occlusion.
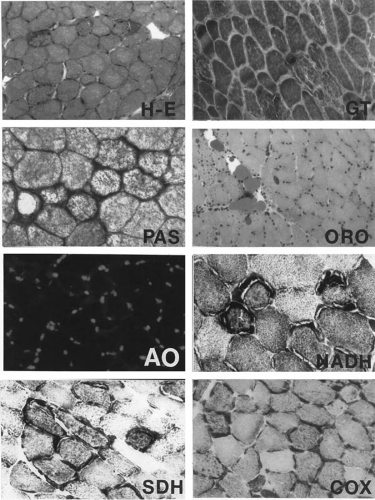 FIGURE 2.2. Quadriceps femoris muscle biopsy of a young woman with right spastic hemiplegia since infancy due to a large left middle cerebral artery infarct and porencephaly. She also has several smaller cerebral infarcts bilaterally, demonstrated by magnetic resonance imaging. She has acquired microcephaly, persistent lactic acidosis, and generalized weakness that has progressed over the last 2 years, hence the muscle biopsy at this time. The clinical diagnosis is MELAS (mitochondrial encephalopathy with lactic acidosis and strokelike episodes) syndrome. Hematoxylin-eosin (H-E) stain shows variation in myofiber diameter and several fibers with basophilic sarcoplasmic masses, corresponding to ragged-red fibers clearly identified with modified Gomori trichrome (GT) stain because these subsarcolemmal zones are irregular in shape and intensely red in color, whereas the normal myofibrils are green. Glycogen is abundantly stained (p-aminosalicylic acid, PAS) in these subsarcolemmal masses and is digested by diastase (not shown); one fiber shows loss of PAS staining except for its ragged-red margins. Neutral lipid, demonstrated by oil red O (ORO), is not increased within myofibers but is globular in the perimysium. Acridine orange (AO) fluorochrome shows no orange-red fluorescence in the ragged-red zones or within myofibrils. The oxidative enzymatic stains nicotinamide adenine dinucleotide (NADH), succinate dehydrogenase (SDH), and cytochrome c oxidase (COX) all exhibit intense mitochondrial enzymatic activity in the subsarcolemmal zones corresponding to ragged-red fibers. Two myofibers show no COX activity and a few others show weak activity, strong evidence of a mitochondrial defect, though nonspecific for which mitochondrial disease. Frozen sections. ×250 (H-E, GT, ORO, AO), ×400 (PAS, NADH, SDH, COX). From a color plate in Sarnat HB, Marín-García J. Pathology of mitochondrial encephalomyopathies. Can J Neurol Sci 2005;32:152–166. With permission of the Canadian Journal of Neurological Sciences. |
Point mutations in mtDNA are the cause of this syndrome; hence the transmission is maternal but not X linked. The most frequent mutation, accounting for about 80% of cases, is an A→G point mutation at the nt-3243 locus, involving the tRNALeu(UUR) gene (35). In another 10% of cases, the locus is T→C at nt 3271 of the same gene. The remaining cases are due to mtDNA point mutations at other loci, and new genetic mutations are being identified continuously; hence MELAS is a true syndrome. The genetic mutation may be identified in blood in most patients; if this approach is not diagnostic, muscle biopsy will identify the ragged-red fiber histopathology and provide tissue for quantitative mitochondrial studies of respiratory chain enzymes, showing a defect in respiratory complexes I and IV and sometimes others as well, and identify known mtDNA point mutations that are screened.
Calcifications in the globus pallidus and other parts of the basal ganglia can sometimes be demonstrated by CT. The MRI appearance and distribution of infarcts large enough to be seen is characteristic (36). Serum creatine kinase (CK) is usually normal, but may be mildly elevated due to an increase in either the BB (brain) or MM (striated muscle) isozyme. Serum lactate is consistently elevated in this disorder, regardless of the specific point mutation and even during asymptomatic phases; pyruvate remains normal or also is elevated. The muscle biopsy is characteristic, as shown in Fig. 2.2 and Table 2.2.
Merrf (Myoclonic Epilepsy with Ragged-Red Fibers)
This condition was first described by Fukuhara and colleagues (37). Clinical onset is during childhood or young adult life, and as the name suggests, the syndrome is highlighted by myoclonus and myoclonic epilepsy, progressive ataxia, and myopathy. Seizures are the usual presenting symptom in childhood and may take mixed forms. Cerebellar ataxia and dementia leading to mental retardation follow and are progressive (38). In contrast to MELAS, cerebral infarcts are uncommon, but some patients have a mixed MELAS/MERRF phenotype. Other, less constant clinical features include small stature, deafness, optic atrophy, progressive external ophthalmoplegia, peripheral neuropathy, and cutaneous lipomas.
Kearns-Sayre Syndrome
First described by Kearns and Sayre in 1958, this ragged-red fiber mitochondrial cytopathy involves brain and muscle (42). It is the most frequent and best studied from both clinical and molecular aspects. The known genetic bases are all different point mutations of mtDNA; hence it is a syndrome, not a single disease.
TABLE 2.2 Mitochondrial Respiratory Chain Enzymes of The Patient Whose Muscle Biopsy Is Shown in Fig. 2.2 | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
As defined by DiMauro and coworkers (43), the condition is characterized by its onset before 15 years but rarely before 5 years of age, the presence of progressive external ophthalmoplegia involving all extraocular muscles, pigmentary degeneration of the retina (retinitis pigmentosa) and one or more of the following: cardiac conduction block, cerebellar deficits, CSF protein greater than 100 mg/dL, or myopathy affecting facial cervical and limb girdle muscles (43). A common additional finding is growth retardation. It can be present before age 5 years and thus precede neurologic and myopathic signs. Peripheral neuropathy occurs in some cases, and altered mitochondria may be demonstrated in axons in sural nerve biopsies (44). Cataracts, neurosensory hearing loss, ichthyosis, a variety of endocrinopathies, and proximal renal tubular acidosis also have been recorded (45,46). The disease is progressive because with time, the fraction of mtDNA containing the deletion increases. This could reflect the fact that mitochondria containing deletions replicate preferentially and accumulate in the ragged-red fibers (47)
Investigations nearly always show a normal serum lactate but elevated CSF lactate. The serum CK is normal or mildly elevated to about 600 IU/L. Visual-evoked potentials often show slow conduction in central visual pathways (geniculocalcarine projections) that is not explained by the retinitis pigmentosa, though visual function usually remains normal except for diplopia resulting from ophthalmoplegia. The muscle biopsy exhibits multiple ragged-red fibers and also increased lipid within myofibers, a feature that helps to distinguish this disorder from
MELAS and MERRF ragged-red syndromes (48). Cardiac investigations should be performed in all patients to detect potentially treatable heart block.
MELAS and MERRF ragged-red syndromes (48). Cardiac investigations should be performed in all patients to detect potentially treatable heart block.
Neuropathologic findings in Kearns-Sayre syndrome include spongiform encephalopathy involving both gray and white matter of the cerebrum and cerebellum and primarily gray matter of the brainstem. Extensive neuronal loss is seen throughout the brainstem and cerebellum, and demyelination of white matter possibly secondary to axonal degeneration. Calcium deposits occur in the thalamus and globus pallidus, involving individual mineralized neurons, and also in the parenchyma. Mitochondrial ultrastructural abnormalities are not consistently shown in the brain despite the neuropathologic lesions (48,49).
Peo (Progressive External Ophthalmoplegia)
This disorder tends to affect adults and rarely has an onset in childhood or adolescence. It is closely related to Kearns-Sayre syndrome both clinically and genetically, and the muscle biopsy also appears virtually identical, including the increased lipid in myofibers, both ragged-red and others (50).
Pearson Syndrome
This ragged-red fiber disorder is marked by refractory sideroblastic anemia, exocrine pancreatic dysfunction, insulin-dependent diabetes mellitus, lactic acidosis, and 3-methylglutaconic aciduria. It is usually fatal during infancy and early childhood, but after age 5 years, survivors develop symptoms and signs typical of Kearns-Sayre syndrome (51). It is clinically distinguishable by the infantile features of endocrinopathies and anemia, the early onset, and the presence of lactic acidosis. The genetic basis is mtDNA deletions similar to those of Kearns-Sayre syndrome, demonstrable in many tissues.
NON–RAGGED-RED FIBER MITOCHONDRIAL SYNDROMES
Leigh Encephalopathy (Subacute Periventricular Necrotizing Encephalopathy)
This syndrome, first described by Leigh in 1951 (52), has many genetic mutations of both mtDNA and nDNA and is the most frequent mitochondrial disease of the perinatal period and early infancy. Transmission may be maternal if the principal mutation affects mtDNA, or it may present as a Mendelian autosomal recessive trait if the principal mutation affects nDNA. X-linked recessive inheritance is a rarer genetic pattern, when a mutation involves the pyruvate dehydrogenase E1 gene on the X chromosome. Autosomal recessive cases often involve pyruvate carboxylase deficiency, SURF1 (a nonencoding “housekeeping” gene of uncertain function) mutation with complex IV deficiency, or isolated complex I deficiency (53,54). A combination of complex III and V deficiency is common, with or without other respiratory chain complexes also being deficient.
Leigh disease can commence at birth. The clinical presentation in the newborn is one of diffuse encephalopathy, but neonatal seizures are infrequent. Infants usually feed poorly or are unable to swallow and may aspirate. Central respiratory insufficiency is sometimes a serious complication in the absence of pulmonary disease and is evidence of brainstem tegmental lesions in the floor of the fourth ventricle involving the fasciculus solitarius. Though the pupils generally remain reactive, the infants do not fixate visually and do not respond reflexively to bright lights shown in the eyes. Reactions to olfactory and auditory stimuli similarly are less than expected. Muscle tone may be hypotonic, or, occasionally, early spastic diplegia may become evident, associated with clonus.
Often infants appear normal at birth, but within the first 3 months of life develop progressive lethargy, visual impairment, dysphagia, and hypotonia with weakness and paucity of movement. Older infants and toddlers may exhibit dyskinesias and progressive cerebellar deficits in addition. In the New York series of Macaya and colleagues, movement disorders, notably dystonia and myoclonus, formed a prominent part of the neurologic picture in 86% of patients (55); in some they were the presenting signs (56). Seizures are uncommon, even in later infancy and early childhood. A male predominance occurs; 66% of patients in the series of DiMauro and colleagues were male (57).
The neurologic deficits are progressive, and most patients die within days to weeks if already symptomatic as neonates or within months to a few years if onset is later in infancy.
In the majority of patients, including neonates, serum lactate is elevated, with normal or mildly high pyruvate. CSF lactate may be elevated despite normal serum lactate. Serum CK is normal. Imaging studies, CT and MRI, show characteristic findings of periventricular leukomalacia and a hyperintense T2 signal in the basal ganglia and thalami. The electroencephalogram (EEG) is usually nonparoxysmal but shows diffuse, poorly regulated, and excessively slow background rhythms in both wakefulness and sleep.
The muscle biopsy is diagnostic even in the neonate (48). Ragged-red fibers are not found (because complexes I and IV are not usually involved together in Leigh encephalopathy and also because these fibers are rare in infancy even in ragged-red fiber diseases); neutral lipid is increased in some cases. Ultrastructural studies of muscle show many abnormal mitochondria with altered cristae. Paracrystalline structures are rare in muscle but are
sometimes seen in the periventricular region of the lateral ventricles at autopsy. Quantitative studies of respiratory chain enzymes in muscle are invariably abnormal. The exact pattern depends on the mtDNA or nDNA defect, which may or may not be evident on screening the common point mutations and deletions. Isolated complex IV (cytochrome c oxidase) deficiency is common, but isolated complex I and isolated complex V or combined complexes III and V as well as pyruvate dehydrogenase deficiencies have also been documented (58,59).
sometimes seen in the periventricular region of the lateral ventricles at autopsy. Quantitative studies of respiratory chain enzymes in muscle are invariably abnormal. The exact pattern depends on the mtDNA or nDNA defect, which may or may not be evident on screening the common point mutations and deletions. Isolated complex IV (cytochrome c oxidase) deficiency is common, but isolated complex I and isolated complex V or combined complexes III and V as well as pyruvate dehydrogenase deficiencies have also been documented (58,59).
Neuropathologic examination shows encephalomalacia and necrosis of periventricular tissue around the lateral ventricles, sometimes the third and floor of the fourth ventricles, and even around the central canal of the spinal cord. Calcified neurons are seen in the thalamus and basal ganglia. Minor developmental malformations are found in the cerebellum, the bases of some cerebellar folia show pan-neuronal loss and gliosis, and convolutions of the inferior olivary nuclei are abnormally formed. Myelination of the white matter of the cerebrum and cerebellum is deficient; small cryptic angiomas of the choroid plexuses may be present (48).
When Leigh encephalopathy is the result of a pyruvate dehydrogenase deficiency, treatment with a ketogenic diet may be beneficial, not only in improving the lactic acidosis and MRI lesions, but in preventing irreversible brain damage (58,59).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


