Chapter 17 Phonation and the production of sound require precise coordination of muscles and consequent airflow and are nearly always perturbed in patients with neuromuscular disorders affecting movement. The neurologic subspecialty of movement disorders has played an increasingly important role in clinical care. Since the last edition of this volume, there has been an explosion of information about genetics, protein chemistry, neurophysiology, and neuropathology. However, with the exception of the major advances in the neurosurgical management of many of these disorders, and in particular the application of high-frequency deep brain stimulation, in addition to the growth in the use of local injections of botulinum toxin as a focal therapy for hyperkinetic disorders, there has been a paucity of specific new pharmacologic interventions. This chapter discusses the fundamental clinical and physiologic characteristics of the more common movement disorders, with special emphasis on the relevant neurolaryngologic aspects. There are several excellent sources for a more detailed general review of movement disorders1,2 in addition to the official journal of the Movement Disorder Society, Movement Disorders. Patients are classified as having a movement disorder if they have a disorder of motor programming resulting in either a paucity of movement (akinesia or bradykinesia), excessive movement (hyperkinesia), or a combination thereof (Table 17.1). In general, for each disorder we classify the disorder as a primary or secondary condition. When the condition is primary, there is no identifiable cause or etiology for the symptoms, which are highlighted when each disorder is discussed.
Movement Disorders of the Larynx
| Movement Disorder | Bradykinetic | Hyperkinetic |
| Parkinsonism | X | |
| Chorea | X | X |
| Essential tremor | X | |
| Dystonia | X | |
| Stuttering | X | |
| Myoclonus | X | |
| Tics (Tourette syndrome) | X |
Proper diagnosis and treatment of these disorders typically requires a team approach including an otolaryngologist, neurologist, and speech-language pathologist. It is helpful to have consultative assistance from a speech-language scientist, psychiatrist, and radiologist. The treating team should be committed to managing these disorders. Once a diagnosis is established, appropriate steps should be taken to appropriately classify symptoms and design a treatment program. Often a treatment program includes treatment of the underlying condition, which may include social support (psychotherapy, family therapy), physical therapies,3 and pharmacotherapy.
For most movement disorders, patients are classified as either idiopathic, that is, without a known etiology or the presence of any other disorder that can present with the phenotype, or symptomatic, that is, with an underlying disorder causing the phenotype. Since the first edition of this book, numerous genetic loci and associated mutations have been identified for cases that historically have been classified as idiopathic or symptomatic, which makes it difficult to classify a patient with genetic underpinnings as “idiopathic”; thus, many authors are classifying patients as “primary” when there is no other underlying neurologic disorder for a disease. It is notable that classifications are typically descriptive of physical signs and have evolved over time as molecular and genetic biology evolve and etiologies become known.4
Parkinson Disease
Definition
Parkinsonism is a neurologic syndrome manifested by any combination of tremor at rest, rigidity, bradykinesia, and loss of postural reflexes.5 At least two of these four cardinal features should be present for the diagnosis of parkinsonism. There are many causes of parkinsonism, and they can be divided into three major categories: idiopathic, symptomatic, and parkinsonism-plus disorders (Table 17.2). The specific diagnosis depends on details of the clinical history, the neurologic examination, and laboratory tests.
|
Abbreviations: Mn, manganese; CO, carbon monoxide; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
Idiopathic parkinsonism, also known as Parkinson disease (PD), is the most common type of parkinsonism encountered by the neurologist. It is a progressive disorder of unknown etiology, and the diagnosis is usually made by excluding known causes of parkinsonism; however, many patients who present with a classical phenotype have a genetic etiology. The major exclusion criteria, which if present indicate another cause of parkinsonism, are presented in Table 17.3 .
|
Source: Fahn S. Disorders with parkinsonism features. In: Hurst JW, ed. Medicine for the Practicing Physician. 2nd ed. Boston: Butterworths; 1988:1522–1525. Adapted with permission.
Clinical Features
Parkinson disease begins insidiously. Tremor is usually the first symptom recognized by the patient; however, the disorder can begin with hypophonia, slowness in movement, or shuffling gait. In the early stages, the symptoms and signs tend to remain on one side of the body, but with time the other side slowly becomes involved as well. Nevertheless, most patients have asymmetric symptoms throughout the course of their disease.
Tremor, approximately 3 Hz, is present in the distal parts of the extremities and the lips while the involved body part is at rest. “Pill-rolling” tremor of the hands is the most typical. The tremor ceases upon active movement of the limb. Resting tremor must be differentiated from postural and kinetic tremors, in which tremor appears only when the arm is being used. These tremors are typically caused by other disorders, namely essential tremor and cerebellar disorders. However, after maintaining outstretched hands for a few seconds, some patients have a reemergence of the rest tremor with the classical approximately 3-Hz frequency.
Bradykinesia is manifested by masked facies; decreased blinking; drooling of saliva due to decreased spontaneous swallowing; loss of spontaneous movement such as gesturing; smallness and slowness of handwriting (micrographia); difficulty with hand dexterity for shaving, brushing teeth, and putting on makeup; short-stepped, shuffling gait with decreased arm swing; and difficulty arising from a chair, getting out of automobiles, and turning in bed. Bradykinesia thus encompasses a loss of automatic movements as well as slowness in initiating movement on command and reduction in amplitude of the voluntary movement. The latter can be observed as decrementing amplitude with repetitive finger tapping or foot tapping.
Rigidity is an increased resistance to passive movement, is equal in all directions, and usually is manifested by a ratchety “give” in the range of motion, so-called cogwheel rigidity. Eventually the patient assumes a stooped posture and begins to lose balance, with a tendency to fall due to loss of postural reflexes. Loss of postural reflexes with stooped posture leads to festination, whereby the patient walks faster and faster.
Onset is usually above the age of 50, but younger patients can be affected, particularly in genetic cases. Onset before age 30 does not preclude a diagnosis of PD, but juvenile parkinsonism raises questions of other etiologies, such as Wilson disease and the Westphal variant of Huntington disease. The disease is more common in men, with a male/female ratio of 3:2. The incidence in the United States is 20 new cases per 100,000 population per year, with a prevalence of 187 cases per 100,000 population.6 With modern medications the mortality rate is close to normal for age.
Pathology and Biochemistry
The pigmented nuclei of the brainstem, namely the pars compacta of the substantia nigra, the locus coeruleus, and the dorsal motor nucleus of the vagus, are affected in PD. There is loss of nerve cells and increased gliosis in these nuclei. The cytoplasm of intact neurons often shows eosinophilic inclusions called Lewy bodies.7
The substantia nigra neurons contain dopamine; the locus coeruleus, norepinephrine; and other pigmented neurons, serotonin. Thus the loss of these neurons results in a loss of these monoamine neurotransmitters in their nerve terminals. The most consistent biochemical alteration in PD is a marked depletion of dopamine in the neostriatum. Most of the symptoms of PD can be directly related to the loss of striatal dopamine. However, some of the motor (e.g., postural instability) and nonmotor (e.g., dementia, depression) features are either incompletely responsive or unresponsive to dopamine replacement therapy, nor is there a halt in the progression of the disease, suggesting that other neurotransmitter systems are involved.8
Historically, environmental factors were deemed to be the primary cause of the condition. Twenty years ago, there was a sparse literature suggesting that PD could have genetic underpinnings,9 and the role of genetics in this condition was hotly debated with the primary example in twin studies. Subsequently, with advances in molecular biology and genetic enabling technologies, including population epidemiology and genetics techniques, nearly a dozen loci have been identified that segregate with the phenotype of idiopathic PD, with additional loci associated with other forms of parkinsonism.10 The current thinking is similar to that recognized by Barbeau11 for which environmental and other factors play upon the neurobiology of appropriately genetically primed individuals and the symptoms of PD emerge. In individual cases, it may be difficult to tease out the relative contributions of nature and nurture. However, the current hypothesis is that the condition is a complex multifactorial neurodegenerative disease whereby the cellular molecular machinery becomes clogged, associated with protein misfolding and subsequent protein aggregation.12 These insights have led to new strategies for both symptom relief and neuroprotection with the hope for more effective therapies in the future.
Speech
In PD, speech production is compromised due to hypokinetic dysarthria with lack of vocal fold closure13 and possibly poor presentation of air to the vocal apparatus (sound generator) due to decreased flow associated with a bradykinetic bellows mechanism. Family members may be first to recognize a change, with patients being unaware. Ultimately, patients complain of hypophonia with a high self-reported voice handicap on formal rating scales such as the Voice Handicap Index.14 The dysphonia, which is often the first speech symptom, is characterized by a decreased loudness with mono-pitch, monoloudness, and prosodic insufficiency. Voice is decreased in loudness and tends to fade out at the end of breath groups, often with a harsh breathy voice quality. Breath groups are shortened and pauses for breaths may occur at inappropriate times. Speech may be produced in short rushes with inappropriate silences between words and syllables. Tremor may be present (discussed later in the chapter). Articulation is produced with reduced range of articulation for both lingual and labial sounds.15,16 Laryngoscopy often reveals bowing of the vocal cords, present to some degree in 87% of patients in one study14 with a midcord opening of the glottis on phonation. The vocal cord motion is often slowed or associated with tremulous movements17 in 53% in one study.14 There may be pooling of secretions in the hypopharynx and difficulties in swallowing. The abnormal movements can be complicated by dyskinesias associated with symptomatic therapy.
Speech is almost invariably affected in multiple system atrophy (MSA), one of the parkinsonism-plus syndromes.18 Autonomic dysfunction is the hallmark of the disease. In parkinsonian type of MSA (MSA-p), formerly known as striatonigral degeneration, the vocal cords may be paralyzed. Ataxic dysarthria is present in cerebellar type of MSA (MSA-c), formerly known as olivopontocerebellar atrophy. There may also be a sensory aberration causing a diminished or absent cough reflex and intermittent aspiration. The respiratory laryngeal function may deteriorate during sleep with stridor and obstructive events, at times life-threatening and result in unanticipated death.19,20 Consequently, a sleep study and appropriate consultations are recommended for all patients who have signs or symptoms of stridor, sleep disturbance, or pulmonary dysfunction. When appropriate, tracheostomy may be required. Surgery may be hazardous and requires close monitoring because of the potential for severe blood pressure fluctuations in MSA patients.
Treatment
A multidimensional approach to treating PD is generally recommended.21 During the past decade, there have been significant advances in the management of PD, including new options for pharmacotherapy, surgical intervention, and speech therapy.
Speech therapy is a primary therapy for PD patients with vocal dysfunction.22 The primary goal is to increase loudness, typically accompanied by increasing the subglottal air pressure, which results in improved vocal fold vibration.23,24 Clinical efficacy has been demonstrated using the Lee Silverman Voice Treatment (LSVT), which has been shown to improve speech and voice measures and vocal fold closure facial expression,25 and lead to changes in neural correlates as seen on neuroimaging.26 However, other treatments are under investigation.27 Speech therapy for PD is discussed in more detail in Chapter 12.
Pharmacotherapy
Patients with idiopathic PD (IPD) typically experience amelioration of symptoms with dopamine precursors (levodopa with carbidopa) either alone or in combination with a catechol-o-methyltransferase inhibitor (entacapone, tolcapone) or dopamine agonists (bromocriptine, pergolide mesylate, ropinirol, pramipexole); however, cases of secondary parkinsonism are typically resistant to medication. Propargylamines28 including selegiline29 and rasagiline,30 have been reported, although not confirmed, to slow progression of IPD if added early in the course of the disease. Their symptomatic effect is mediated through inhibition of monoamine oxidase type B enzyme. Select dopamine agonists have been suggested to slow progression of IPD. However, because of the methodologic challenges in distinguishing symptomatic benefit from delayed neurodegeneration, conclusive data are not yet available.31,32
Long-term levodopa therapy often leads to late complications, including response oscillations, dyskinesias, and drug-induced psychiatric effects. Early in therapy, most patients have continuous benefit from three or four doses per day; subsequent response oscillations typically develop within 5 years of treatment. Even more troubling, some patients develop unpredictable oscillations with no apparent relation to their dosing regimen. Response oscillations may be accompanied by dyskinesias; chorea, with or without dystonia, is common, especially at the time of peak levels. Although dose reduction may be effective for peak-dose dyskinesias, many patients have a very narrow therapeutic window, and titrating an effective dose versus reduction of the dyskinesia may be difficult. Strategies include sustained-release preparations, coadministration of dopamine agonists, liquid preparations, metabolic inhibitors such as catechol-o-methyltransferase inhibitors, or subcutaneous apomorphine. Delayed gastric emptying may be responsible in some cases. Cisapride33 had been used in select patients in the past to improve gastric motility but was withdrawn from the U.S. market due to its cardiac side effects. Psychiatric effects of chronic levodopa therapy include vivid dreams and hallucinations, and an exacerbation of memory loss, confusion, and anxiety. Although some of these effects may be treated symptomatically8 with other medications, a reduction in levodopa may be necessary.
Oral therapies that result in enhanced stimulation of dopamine receptors (e.g., levodopa, dopamine agonists, dopamine releasers) and an overall improvement in motor function have been clearly demonstrated to improve speech production in general.34 Although paradoxical, many of the characteristics of speech, including respiration, phonation, and articulation may be perturbed by levodopa-related fluctuations. In addition, pharmacologic treatment can sometimes worsen speech by making it more rapid, less distinct, with some “freezing of speech,” that is, repetition of a syllable or phrases. Vocal cord augmentation35 has been used to correct the open glottis on phonation, but the results may be poor if there is a significant reduction in air presented to the larynx. Therefore, pulmonary function studies are important before augmentation.
Neurosurgery
In the early 20th century, lesions of the motor cortex were performed in an effort to ameliorate hyperkinetic movements, including tremor associated with PD.36 However, these excisional procedures in addition to incising the pyramidal tracts in the upper cervical cord often resulted in a spastic hemiparesis as a substitute for the dyskinesia. In 1940, Meyers37 described pallidotomy for PD, later demonstrating that dyskinesias can be improved with his procedure without pyramidal tract damage.38 During a surgical procedure in a postencephalitic PD patient, Cooper39 accidentally severed the anterior choroidal artery, which feeds the medial globus pallidus and lateral ventral thalamus, and discovered marked improvement in tremor and rigidity. Cooper40 later reported that dystonic posturing in parkinsonian patients improved with anterior choroidal artery ligation, and he began to operate on primary dystonia patients. Cooper performed lesions in the pallidum and thalamus with improved tremor control.41 With bilateral procedures, there was a significant morbidity due to dysarthria and dysphagia. Once levodopa became available in the late 1960s, surgical interventions were rarely performed until recent decades.
During the past two decades, there has been a resurgence of interest in surgical interventions for PD prompted by several factors. Chronic levodopa therapy typically results in motor fluctuations, which emerge within the first 3 to 5 years of therapy. Strategies have been developed to attempt to delay the onset of motor fluctuations, but for most patients these complications ultimately emerge. Technical advancements in neuroimaging, stereotactic neurosurgery, and microelectrode intraoperative recording have permitted improved localization of surgical targets and placement of either lesions or implants. Finally, our understanding of the organization of the basal ganglia and those pathways important to the pathophysiology of PD has led to a more rational approach to target selection.42
Neurosurgical procedures for PD can be broadly classified into (1) ablative procedures, (2) stimulation procedures, and (3) potentially restorative procedures. Detailed reviews are available.43
Ablative procedures have been performed in the thalamus and internal segment of the pallidum. Thalamotomy can be performed with modest morbidity, and has demonstrated improvement in contralateral tremor and, to a lesser extent, in rigidity, in up to 90% of patients.44 In most cases, thalamotomy does not result in improvement for bradykinesia, postural instability, or ipsilateral tremor. Currently, mortality is less than 1%44; complications include contralateral hemiparesis, seizures, paresthesias, ataxia, apraxia, hypotonia, abulia, and gait disturbances. Pallidotomy gained attention45 because when the lesion is placed in the posteroventral portion of the internal segment of the globus pallidus (GPi), there can be long-lasting improvement in tremor, rigidity, hypokinesia, speech, gait, dystonia, and, importantly, levodopa-induced dyskinesias. Utilizing microelectrode recordings for target localization, many groups have reported impressive and dramatic improvements in dyskinesias with more modest improvements in the cardinal signs of PD.46
Deep brain stimulation (DBS) for the management of PD was introduced by Benabid and colleagues.47 They showed that high-frequency (>100 Hz), low-voltage (<5 V) electrical stimulation of the ventro-intermediate (Vim) nucleus of the thalamus improved tremor in PD and essential tremor. Based on a growing understanding of the neural networks controlling basal ganglia function in PD and other disorders, attention shifted to stimulating the GPi, and subthalamic nucleus (STN) in patients with intractable PD. Early trials48 demonstrated that in addition to improvement in tremor, the other cardinal features of PD, namely bradykinesia, gait disturbance, rigidity, and balance, may be improved by DBS of the GPi or STN. This has translated into an improvement in activities of daily living (ADL) scores and motor function.49,50
Transplantation is an experimental procedure whereby dopamine-containing cells are transplanted into the nigral targets of patients with intractable PD. The foundation for this therapy is based on our understanding that PD is associated with specific degeneration of the dopaminergic nigrostriatal neurons, and dopaminergic replacement strategies result in improvement of the motor signs and symptoms of PD. There are numerous factors to consider when considering transplantation therapy, including choice of tissue to be transplanted (adrenal, fetal nigral, retinal pigment epithelial, stem cells, etc.), density and quantity of tissue to be transplanted, location of transplant, and the role of immunosuppressive therapy. Transplantation of adrenal medullary cells into the caudate nucleus was initially reported by Madrazo et al51 to have dramatic results, but the results were not reproduced by other investigators52,53; the procedure has been abandoned. Early open-label studies of fetal nigral cell transplantation into the posterior putamen demonstrated feasibility, improvement in symptoms, and improved striatal uptake in levodopa.54,55 Postmortem studies demonstrate that with at least some transplant protocols, implanted cells can survive in large numbers and reinnervate the striatum in an organotypic fashion.56 In grafted regions, there is normal-appearing staining for dopamine transporter and cytochrome oxidase, normal tyrosine hydroxylase (TH) messenger RNA (mRNA) expression, and normal-appearing synaptic connections between grafted and host neurons without evidence of host-derived sprouting.57 Two double-blind studies of fetal nigral transplantation, supported by the National Institutes of Health, have been reported. The first study, reported by Freed et al58 in 2001 demonstrated modest benefit in the younger patients (under age 60). No improvement was derived from the implant despite neuron-imaging evidence that the graft survived and produced dopamine. However, there was a marked placebo effect and some patients developed unanticipated severe dyskinesias when not on medication that could not be ameliorated by decreasing dopamine therapy.59 The second study performed under a different transplantation protocol showed no overall treatment effect, but milder patients did show significant improvement.60 Neuroimaging demonstrated transplant survival, and postmortem examination showed robust survival of dopamine neurons. Although neuroimaging and immunohistochemistry supports the potential role of neuro-transplantation, the technology is not considered accepted at this time.
Recently, investigators have reported speech and intelligibility benefits of DBS of the subthalamic nucleus when used to treat PD. One study reported mild improvement, particularly for modulation in loudness and pitch, at the expense of intelligibility.61 Others have reported improvement in perceptual speech as documented on global PD rating scales and quantitative acoustic and force analyses.62 However, not all authors have reported benefits from DBS at this site.63–67 The benefits from DBS are dependent on an anatomic substrate of speech dysfunction in PD, the pharmacologic status of the patient, and stimulation location and parameters. Pinto et al68 examined four patients under a variety of conditions and concluded that motor speech subcomponents can be improved similar to limb motor function, but that the complex coordination of all speech anatomic substrates are not uniformly responsive to subthalamic nucleus stimulation. It should also be noted that brain surgery can contribute to speech disorders by impacting the motor and language circuits, and this must be taken into consideration when evaluating a patient who has had central nervous system (CNS) surgery.69
Dystonia
Dystonia is a syndrome dominated by sustained muscle contractions frequently causing twisting and repetitive movements or abnormal postures that may be sustained or intermittent. Dystonia can involve any voluntary muscle. Because the movements and resulting postures are often unusual and the condition is rare, it is one of the most frequently misdiagnosed neurologic conditions.70 The prevalence estimates of dystonia suggest approximately 50,000 to 200,000 cases of idiopathic dystonia in the United States.71 Additional epidemiologic studies have shown the following: a service-based prevalence of primary focal dystonia of 13.7/100,000 in the Tottori Prefecture of Japan72 and a crude prevalence for all types of primary dystonia of 37.1/100,000 and laryngeal dystonia of 5.9/100,000 in Iceland.73 During the past decade, major advances have occurred in the understanding of the genetics and biology of the condition, in addition to providing further support for therapeutic management with botulinum toxin and DBS.
As a clinical syndrome, patients can be classified according to clinical symptomatology, age at onset, and etiology. Classification may be important as it can give us clues about prognosis and also an approach to management. The classification scheme is outlined in Table 17.4 .
|
Source: Data from Fahn,501 Calne,502 and Bressman.503 Those reviews should be consulted for references regarding the literature citations for these etiologies.
Dystonia may begin at any age. We have seen presenting signs as early as 9 months and as late as 85 years. In general, there is a bimodal age-at-onset distribution with peaks at ages 8 and 42 (Fig. 17.1). Therefore, we classify patients as early onset when the presenting signs are before age 26, and as late onset at older ages. Alternatively, we can classify patients as infantile onset when the presenting signs are before age 2, childhood onset between ages 2 and 12, adolescent onset between ages 13 and 20, and adult onset at older ages. The significance of this distinction is discussed below.
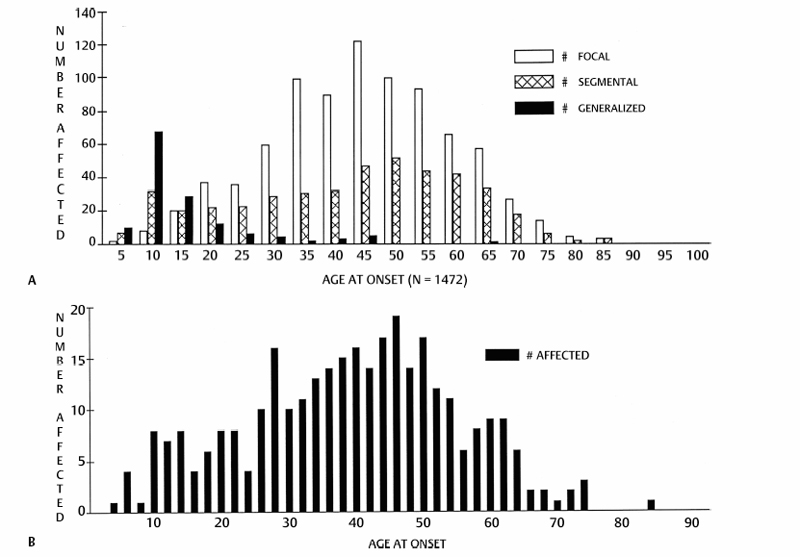
Fig. 17.1 Age at onset distribution of patients with primary dystonia evaluated at the Columbia University Dystonia Clinical Research Center as of 1990. (A) All patients with primary dystonia. (B) All patients with primary laryngeal dystonia.
According to the etiologic classification, patients with idiopathic disease have no evidence by history, examination, or laboratory studies of any identifiable cause for the dystonic symptoms. Therefore, there must be a normal perinatal and early developmental history; no prior history of neurologic illness or exposure to drugs known to cause acquired dystonia (e.g., phenothiazines); normal intellectual, pyramidal, cerebellar, and sensory examinations; and normal diagnostic studies. Patients who have abnormalities noted above are classified as having secondary dystonia. The clinical phenomenology will often be a clue as to etiology. Primary dystonia is typically action-induced; symptoms are enhanced with use of the affected body part, and the region may appear normal at rest. Secondary dystonia frequently results in fixed dystonic postures. The presence of extensive dystonia limited to one side of the body (hemidystonia) suggests a secondary etiology.
When classified by the distribution, patients are categorized as having focal, segmental, or generalized symptoms. Focal dystonia symptoms involve one small group of muscles in one body part, segmental disease involves a contiguous group of muscles, and generalized dystonia is widespread. Dystonia may involve muscles of the oral cavity, larynx, pharynx, tongue, and jaw. Common examples of focal dystonia (Table 17.4) include blepharospasm (forced, involuntary eye closure), oromandibular dystonia (face, jaw, or tongue), torticollis (neck), writer’s cramp (action-induced dystonic contraction of hand muscles), and spasmodic dysphonia (vocal cords).
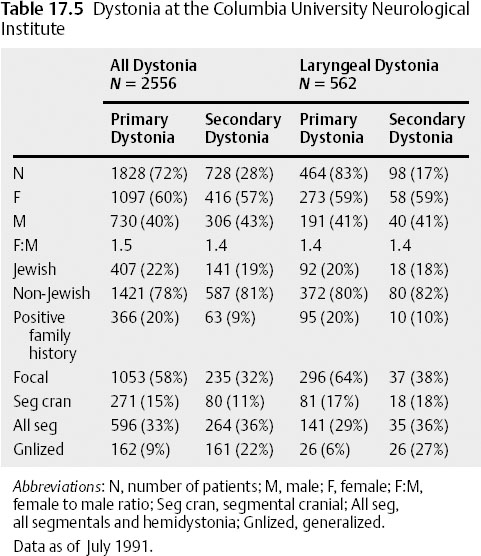
Idiopathic and hereditary generalized dystonia almost always begins as a focal dystonia before spreading to involve other parts of the body. Spread may not take place or may be limited and then plateau as a focal or segmental dystonia. Age at onset, genetic predisposition, or other factors likely contribute to the phenotypic expression in any particular individual. The coexistence of dystonic symptoms affecting the vocal cords and other body parts further supports the notion that the laryngeal symptoms are dystonic, as discussed below (Table 17.5).
Spasmodic Dysphonia (Laryngeal Dystonia)
Idiopathic spasmodic dysphonia (SD) and laryngeal dystonia (LD) are clinical terms used to describe an action-induced, laryngeal motion disorder. Most cases represent manifestations of primary dystonia, but many are secondary to other neurologic entities (Table 17.4).
In 1871, Traube74 coined the term spastic dysphonia when describing a patient with nervous hoarseness. Schnitzler et al75 used the terms spastic aphonia and phonic laryngeal spasm to describe such patients. Gerhardt76 called the condition “coordinated laryngeal spasm.” Fraenkel77,78 uses the word mogiphonia for a slowly developing disorder of the voice characterized by increasing vocal fatigue, spasmodic constriction of throat muscles, and pain around the larynx. Of note, he compared the laryngeal disorder to “mogigraphia” or occupational writer’s cramp, or dystonic cramping of the arm when writing. In 1899, Gowers79 described functional laryngeal spasm whereby the cords were brought together too forcibly while speaking (currently classified as adductor type). He contrasted this to phonic paralysis, whereby the vocal cords could not be brought together while speaking (currently classified as abductor type). Gowers wrote, “The affection has been compared to writer’s cramp… a case reported by Gerhardt, in which the patient had actually suffered from writers’ cramp, and, at the age of 50, learned to play the flute. The act of blowing the flute brought on laryngeal spasm and an unintended voice sound, accompanied by muscular contractions in the arm and angle of the mouth.” Here again, the focal dystonia of LD is being compared with dystonia involving other segments of the body (mouth, arm). Critchley80 described the voice pattern as a condition in which the patient sounds as though he were “trying to talk whilst being choked.” Bellussi81 described the condition as “stuttering with the vocal cords.”
Aronson drew specific attention to this disorder in the laryngeal literature. At the time of his 1968 review,82 there were approximately 122 cases in the literature, including 34 of his own cases. Minnesota Multiphasic Personality Inventory (MMPI) testing and psychiatric interviews did not discriminate between patients with LD and those in the normal population, distinguishing those affected with LD from patients with psychogenic dysphonias, thus helping to establish SD as an organic, or nonpsychiatric, condition. Nevertheless, many patients are referred to psychiatrists for treatment because the correct diagnosis is not made when the patient initially presents for treatment.
Aronson83 later formally distinguished and reviewed two types of spasmodic dysphonia: adductor, due to irregular hyperadduction of the vocal folds; and abductor, due to intermittent abduction of the vocal folds. Patients with adductor LD exhibit a choked, strained-strangled voice quality with abrupt initiation and termination of voicing, resulting in short breaks in phonation. The voice is generally reduced in loudness and monotonal. Vocal tremor is frequently observed along with a slow speech rate and decreased smoothness of speech. Speech intelligibility is generally decreased. Occasionally patients with adductor LD exhibit compensatory pseudo-abductor spasmodic dysphonia, compensating for severe adductor laryngeal spasms by abducting their vocal cords and whispering. Patients with abductor LD exhibit a breathy, effortful, voice quality with abrupt termination of voicing, resulting in aphonic whispered segments of speech. The voice is reduced in loudness and vocal tremor is frequently observed. Speech intelligibility is generally reduced. Some patients display a combination of adductor and abductor signs and have been classified as mixed.
Aronson and Hartman84 later noted that LD has tremor characteristics similar to those found in essential tremor (ET). The differential diagnosis between LD due to ET and that due to a dystonic tremor can be difficult. We noted that spasmodic dysphonia is not a spastic disorder85; electromyographic characteristics were inconsistent with those seen in pyramidal disorders. We found an irregular tremor in 25% of patients as opposed to the regular tremor of essential tremor (discussed below). The findings were comparable to those of patients who had generalized dystonia with laryngeal involvement. Schaefer et al86 used electromyography to establish further that LD is a disorder of vocal motor control. Because the condition is not a spastic disorder, we had favored the term spasmodic dysphonia rather than spastic dysphonia.
Several lines of evidence further supported the notion that spasmodic dysphonia is a form of dystonia. Fraenkel77,78 and Gowers79 compared the involuntary movements in spasmodic dysphonia to those of other dystonias. However, in the earlier works, a psychogenic etiology was proposed.87 Jacome and Yanez88 associated spasmodic dysphonia with Meige disease, or segmental-cranial dystonia (Table 17.4); other authors subsequently have concurred.89–92 Many of the clinical and electrophysiologic phenomenologic and genetic features of patients with focal spasmodic dysphonia are similar to those with more generalized disease (see below), and we have recommended that the condition be properly called “laryngeal dystonia.”
Furthermore, dystonia is characterized by abnormal involuntary movements that are typically action induced. In LD, the action is that of speaking. The vocal apparatus is usually normal at rest, but functions abnormally with speaking. Adductor LD is characterized by abnormal involuntary cocontraction of the vocalis muscle complex muscles resulting in inappropriate adduction of the vocal folds. As an action-induced, task-specific, or functional movement disorder, the muscles and anatomic structures are typically normal at rest but move inappropriately with action. Conversely, abductor LD is characterized by action-induced inappropriate cocontraction of the posterior cricoarytenoid muscles during the action of speaking, resulting in inappropriate abduction of the vocal cords. Patients with mixed adductor and abductor dysphonia have both prominent adductor and abductor spasms.93
We have reported patients with respiratory adductor laryngeal dystonia.94–97 These patients have abnormal involuntary adduction of the vocal cords on respiration, but may have grossly normal function with speech. In this case, the specific action is the movements of the vocal cords during respiration. Patients may be idiopathic or have a tardive etiology. Symptoms may be laryngeal at onset or begin in other cranial regions (upper face as blepharospasm) and progress to involve the larynx and other cranially innervated structures (e.g., jaw, diaphragm). Laryngeal stridor was relieved with local injections of botulinum toxin in some of our cases whereby the respiratory dysrhythmia persisted. Furthermore, we have also reported cases whereby the laryngeal hyperkinesias were induced by the action of singing,98 with normal vocal cord function and connected speech at initial presentation. It is notable that whereas singing provoked dystonic symptoms in these patients, we have also treated cases of cervical dystonia (CD) and blepharospasm whereby singing was a trick used to temporarily improve dystonic symptoms.
Dystonic movements can be rapid and repetitive and tremor may be seen in dystonia affecting any segment of the body. Dystonic tremors are typically irregular and have a directional preponderance; symptoms are increased when the patient’s posture places the affected body part in a position opposed to the primary dystonic contractions. For instance, patients with torticollis often have a head tremor that can be damped by placing the head into the preferred posture. Many patients with SD have an irregular vocal tremor that is both audible and can be recorded electromyographically.86 Similar to the dystonic tremor seen in individuals with arm or neck dystonia, the irregular dystonic tremor of SD may be due to posturing of dystonic muscles in a position where the agonist contractions do not fully neutralize those of the antagonists. This tremor may be differentiated from the regular tremor seen in benign essential voice tremor (see Tremor, later in this chapter). The clinical distinction may be difficult in many cases, particularly when a patient presents with symptoms of essential tremor in other body parts. In many cases, the clinical distinction cannot be made.
Sensory System and Dystonia
Sensory tricks often ameliorate dystonic movements and postures, and this maneuver can be effective in different parts of the body. These sensory tricks are also known as a geste antagonistique or gegendruckphanomen. The patients with CD often find that gently touching the chin, back of head, or top of head relieves the symptoms; blepharospasm patients may relieve symptoms by gently touching the lateral canthus. The use of the sensory tricks to keep the head in the body midline position was reported by 88.9% of patients in one series.99 The physiology of sensory tricks remains unknown. In a recent study, 13 of 25 patients with idiopathic CD had markedly reduced electromyography (EMG) activity (50% or more) even during arm movement, but before the arm touched the skin, while performing a sensory trick.100 Some patients have been reported to have reduced dystonia while thinking about a sensory trick.101 Patients with SD have reported that symptoms momentarily improve by pinching the nares, pressing the hand against the back of the head,102 pressing the hand into the abdomen, pulling on an ear, or touching the clavicular notch (personal experience). Many patients observe that they speak better after a yawn or sneeze, or when they sing or yell; these are common experiences in patients with other cranio-cervical dystonias and were reported in early descriptions of dystonia.103,104
Botulinum toxin type A (BONTA) may also modify the sensory feedback loop to the CNS, and this mechanism may be partially responsible for its beneficial effect in treating dystonia. Ludlow et al105 and Zwirner et al106 proposed that reduced muscle activity and therefore feedback to laryngeal motoneuron pools may be a primary mechanism of action of BONTA. We offered the possibility that toxin might have a direct effect on sensory afferents by blocking intrafusal fibers, resulting in decreased activation of muscle spindles.107 This would effectively change the sensory afferent system by reducing the Ia traffic. Filippi et al108 supported this hypothesis by establishing that local injections of BONTA directly reduce afferent Ia fiber traffic, and therefore exert a modulatory effect on sensory feedback. This may also account for the clinical observation that injections of BONTA have an effect on regional noninjected muscles, which is most striking in spastic limbs.109
Support for this mechanism derives from the cumulative work of Ryuji Kaji and colleagues,110–115 who showed that the increase in severity of dystonic writer’s cramp associated with enhancing Ia muscle spindle activity with the tonic vibration maneuver can be decreased by intramuscular injections of dilute lidocaine, which preferentially affects the afferent innervation of the muscle spindle. Both ethanol and lidocaine block sodium channels; however, the former blocks the channels for a longer duration than the anesthetic. Kaji et al called the treatment of lidocaine plus ethanol the “muscle afferent block” (MAB), and it has shown an effect in neck, jaw,114 and limb dystonia110,111 and spasticity.113 ,115 The benefit for each treatment lasts only a few weeks, and therefore is of limited use in most dystonic and spastic situations. However, this model of blocking Ia afferents supports the proposed mechanism of action with BONTA on conditions associated with excessive muscle contraction. These studies support the importance of the afferents system in the clinical manifestations of dystonia.116
Also notable is the growing appreciation of the importance of the integrative sensory function of the basal ganglia as reviewed by Kaji’s group117,118 and recently DeLong and Wichmann.42 In addition to the observations described above in patients with dystonia who can respond to a sensory or tactile stimulus, PD patients have demonstrated a remarkable ability to have enhanced motor function as a consequence of specific sensory cues, the phenomenon of kinesie paradoxale 119 (as discussed by Riess and Weghorst120). For instance, PD patients in the “off” state may be able to run out of a room in response to an emergency, such as someone yelling “fire,” or walk more effectively on a sandy beach rather than on a floor. Sensory cues, such as stepping over lines on a floor, have been used to facilitate gait in PD patients. These observations, coupled with the remarkable understanding of the basal ganglia stimulated by advances in cellular recording during DBS surgery, have supported the notion that the basal ganglia are an important relay for the gating of both sensory and motor circuits whereby sensory inputs have the ability to control motor output.
In patients with cranial dystonia, involuntary hissing or humming may be present. It is difficult to know if it is primarily associated with the disorder or is a secondary trick to relieve the patient of symptoms.121 We have observed numerous patients with laryngeal dystonia who hum before speaking, as if to initiate vibration of the vocal cords to prepare them for vocalization.
Trauma has become generally accepted as a factor that may trigger dystonic symptoms,122–124 although this matter is debated by some.125 Head/neck trauma has been reported in 5 to 21% of cervical dystonia cases.126–129 Patients with spasmodic dysphonia may report the onset of symptoms immediately after a laryngeal/pharyngeal trauma, most often following a viral infection130,131; this observation has been borne out in our series of patients. Whereas up to approximately 30% of our patients with dystonia have a clear family history of dystonia, and therefore a genetic predisposition to the development of symptoms, some of the sporadic cases may be genetically susceptible or “primed,” and after exposure to the appropriate environmental factors (“trigger factors”) such as exposure to infections or trauma, symptoms manifest. Limb dystonia132,133 and jaw dystonia134 may occur after peripheral traumas. Similar models have been proposed for other movement disorders.11 There is a growing body of experimental evidence that the appropriately predisposed may develop dystonia,135,136 including with overuse/overactivity.137 In our practice, we accepted trauma as a trigger for dystonia when the onset of the dystonia is within 6 to 12 months of the identified trauma. In many cases, the peripheral injury that preceded the dystonia was acute, brief, and well defined. In some of our patients, the injury was relatively mild or chronic or repetitive, as had been noted by Schott.138 The dystonia typically occurs in the traumatized body part or region, and in many cases is associated with pain. Sometimes the dystonic posture evolves as the pain improves.
Psychogenic Dystonia
We consider psychogenic dystonia to be a form of secondary dystonia because the phenomenology is dystonic and the etiology is psychiatric. Suspicious findings on neurologic examination suggestive of a psychiatric etiology include false weakness, false sensory complaints, multiple somatizations, obvious psychiatric disturbances such as self-inflicted injuries, incongruous and inconsistent movements and postures, and distractability. Some patients have improvement with suggestion, or unusual and nonphysiologic intervention. Psychogenic dystonia can be severe enough to lead to fixed contractures. Fahn and Williams139 proposed a classification scheme, stating that the clinical diagnosis is made with certainty only after symptoms improve without pharmacotherapy in the setting of a history and examination that are inconsistent or incongruous with an organic movement disorder.140–142 Improvement with a gestes antagoniste is uncommon but has been reported.143 ,144
However, many patients with unusual movement disorders are misdiagnosed as psychogenic because of the unusual nature of their symptoms and signs. Stanley Fahn and the late David Marsden stated, “Just because it is unusual does not mean it is psychogenic” (personal communication; also see Shale et al145 ), emphasizing that the diagnosis of a psychogenic movement disorder is complicated because the symptomatology often mimics that of other neurologic disorders.146 These patients can be extraordinarily difficult to diagnose. For instance, the diagnosis of psychogenic dystonia was made in a patient with the classical DYT1 gene mutation in a family with dystonia.147,148 Some patients may present with fixed dystonic postures144,149; neurophysiologic abnormalities, consistent with organic dystonia, have been observed in psychogenic dystonia.142,150 However, the consequences of misdiagnosis, whereby psychogenic patients are classified with an organic movement disorder, and vice versa, can be significant. The patient with organic disease can be subjected to years of demoralization and psychotherapy associated with a diagnosis of an unreal disorder, whereas patients with psychogenic disease can undergo unnecessary treatments, including neurosurgical procedures,142,151 with all good intentions, when they are diagnosed as having an organic etiology for their symptoms. A rating scale has recently been developed for understanding the phenomenology of patients with psychogenic movement disorders.152 Although many patients remain chronic, a multidisciplinary approach including a focus on psychiatric intervention, can be successful.
In patients with LD, clinical features, such as improvement with alcohol, sedatives, and tranquilizers, and worsening under stress or when talking on the telephone, had been used as evidence that patients presenting with symptoms of LD have a psychogenic basis for their condition. However, these are common clinical features among the dystonias. Similar to torticollis,153,154 remissions may occur in patients with LD, and some relapse.
We suspect that psychogenic LD is very rare, but the true incidence is not known. Speech language pathologists have described cases to us, and we suspect that we have not seen these patients because they are treated by speech-language pathologists and are not referred for evaluation. One of our patients had a very bizarre speech pattern that was thought to be psychogenic. With speech therapy, the bizarre speech resolved, and she was left with pure abductor LD. In retrospect, the bizarre speech was a manifestation of futile attempts to develop a compensatory strategy.
Laryngeal tension-fatigue syndrome and muscle tension dysphonia are terms used to describe secondary muscle tension disorders. The laryngeal, perilaryngeal, suprahyoid, neck, and jaw muscles may be involved.155,156 Increased muscular tension in the neck and larynx has been found to be associated with palpable increased muscle tension in the suprahyoid and perilaryngeal muscles during phonation, elevation of the larynx with increased pitch, open posterior glottic chink on phonation, and associated vocal fold abnormalities such as nodules or chronic laryngitis.157–159 Under mild conditions, alterations in pitch and easy fatigue may be present, and chronic intermittent dysphonia may occur. With more significant musculotension dysfunction, the voice may become breathy and harsh to variable degrees.160 These vocal presentations are sometimes difficult to differentiate from spasmodic dysphonia.161–163 Such muscular tension-related disorders are often noted in professional speakers. Relaxation techniques, appropriate neck and shoulder positioning, and instruction for efficient voice use is recommended. Nonsteroidal antiinflammatory drugs (NSAIDs), physical therapy, warm compresses, and massage may also help.164 Forced whispering can result in muscle tension dysphonia (MTD).
Genetics
Idiopathic torsion dystonia (ITD) is thought to be etiologically heterogeneous and clinical and ethnic subtypes of ITD have been identified during the past three decades of focus. When the Dystonia Clinical Research Center in New York was funded in the early 1980s, the primary objective was to identify the causes of dystonia and develop effective therapies. At that time, based on our observations,165 we thought that the age at onset of dystonia might provide some clues to the underlying pathobiology of the condition. The age-at-onset histogram with its bimodal distribution did indeed provide a window into the genetics, predicting that most patients with childhood-onset dystonia would have a different genetic etiology than those with adult-onset dystonia.165 We also knew from the analysis of pedigrees that even in families that had a clear autosomal dominant pattern of inheritance, there was reduced penetrance whereby not all gene carriers manifested symptoms. Family studies suggested that different phenotypes likely indicated a different genetic basis for the observed clinical heterogeneity.
The initial focus of our research was to understand the genetics of one of the most severe forms of inherited dystonia, childhood-onset idiopathic dystonia, also known as primary generalized torsion dystonia (PTD), dystonia musculorum deformans, or Oppenheim’s dystonia.166 It is inherited as an autosomal dominant disease with a 30 to 40% penetrance.166 The symptoms usually start in an arm or leg in childhood, and generalize by adulthood. Occasionally onset is in later life, with the symptoms typically presenting in the cranial structures (e.g., neck, larynx, and upper face), and tending to stay more localized.167 The cause for this variation in distribution with different age of onset is not known, but likely reflects the underlying neurobiology of the condition168 caused by the DYT1 gene deletion mutation, which was identified in 1997. Although the deletion mutation is most prevalent in Ashkenazi Jews due to a founder mutation,166 it can appear in most ethnic populations, including African-Americans and Asians. In some cases the deletion appears as a spontaneous mutation.169,170
Extensive work on DYT1 dystonia led to the identification of torsinA, the protein product of the DYT1 gene, and its localization to chromosome 9q34.1.171–174 In most pedigrees with the clinical syndrome of childhood-onset PTD, there is a single amino acid (glutamic acid) deletion at residue 303.174 This alteration in the amino acid sequence of the protein is necessary, but not sufficient, for the production of the clinical symptomatology, as not all gene carriers develop symptoms of dystonia. The reason for the lack of complete penetrance remains to be elucidated, but may be attributable to some property of the abnormal torsinA in dystonia, for example, the precipitation of neuronal dysfunction by an environmental factor(s) or by other modifier genes, small interfering RNAs or other transcriptional factors, resulting in alterations of gene-protein interactions.
The role of torsinA in cellular function has not yet been fully elucidated. Based on the amino acid sequence, torsinA has an adenosine triphosphate (ATP) binding domain, and has a sequence that is similar to that of the heat shock proteins (HSPs). HSPs have ATP and adenosine triphosphatase (ATPase) activity, and they have chaperone functions. Mutations in the carboxy region of these proteins can block binding to companion proteins. Therefore, if native torsinA functions in a multimer, then a mutation could result in disassociation, which is characteristic of dominant-negative disorders and manifest as a dominant disorder. Studies have demonstrated that the mutant torsinA that is normally localized in the endoplasmic reticulum relocates to the nuclear envelope, and potentially brings the wild-type torsinA with it, thus disrupting normal cellular function.175–178
Although the initial focus had been on DYT1 dystonia, additional genetic subtypes, distinguished by phenotype, molecular biology, or genotype, have been identified and are summarized elsewhere.179 Hereditary autosomal dominant myoclonus dystonia is a movement disorder characterized by involuntary lightning myoclonic jerks of the axial and appendicular structures, and mild dystonic movements and postures are typically alleviated by alcohol. We reported a family with eight members with myoclonus dystonia180 ; the index case presented with a diagnosis of laryngeal dystonia. Using a positional cloning approach, Zimprich et al181 identified five different heterozygous loss-of-function mutations in the gene for varepsilon-sarcoglycan (SGCE), in patients with myoclonus dystonia. Pedigree analysis showed a marked difference in penetrance depending on the parental origin of the disease allele, indicative of a maternal imprinting mechanism, similar to that which has been demonstrated in the mouse varepsilon-sarcoglycan gene. It is interesting that many of these patients have associated obsessive-compulsive disorder (OCD) and alcohol dependence.182 This is reflected in the neuropsychiatric manifestations and alterations in dopaminergic and serotonergic metabolites in an animal model of this condition.183
Mutations in the mitochrondrial genome can also result in dystonic signs. In addition to dystonia, these patients typically display additional features associated with mitochondrial dysfunction, such as optic atrophy, hearing loss, peripheral and neuropathy. In some cases abnormal neuroimaging provides diagnostic clues.184
In most families with dystonia, various members may have different presentations. For instance, in DYT1 dystonia, individuals may present with generalized, segmental, or focal dystonia, suggesting that those with milder manifestations are related to the more generalized cases. This observation supports the concept that the less involved individuals are formes frustes of generalized dystonia. Across all forms of dystonia with laryngeal involvement, the presence of dystonic symptoms in other family members supports the concept that the laryngeal involvement is dystonic and genetic in these cases (Fig. 17.2). For instance in our series of patients, 11 to 31% of patients presenting for evaluation and treatment of laryngeal dystonia had other family members with dystonic symptoms involving the larynx or some other segment of the body.18 5 Those with a family history of dystonia in general had a younger age at onset.
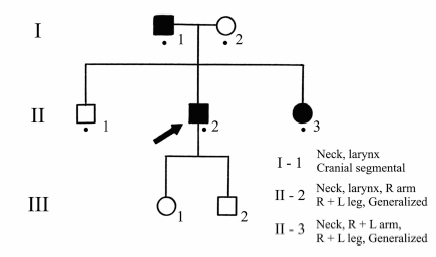
Fig. 17.2 Family C. The proband has generalized dystonia with vocal cord involvement. His sister has generalized dystonia with prominent torticollis. Their father has involvement of his neck and vocal cords.
Laboratory Evaluation
Routine laboratory investigations are typically normal in patients with idiopathic dystonia, including idiopathic SD. Patients with secondary dystonia have laboratory findings consistent with the underlying disorder. Neuroimaging studies with positron emission tomography (PET) have suggested abnormalities in the basal ganglia and associated outflow pathways as reflected in the sensory186,187 or motor18 8 cortex in classical idiopathic disease, and motor cortex189 in X-linked dystonia-parkinsonism (“Lubag”190,191). Eidelberg et al192 has studied clinically nonmanifesting and manifesting DYT1 carriers and found that they express a specific metabolic topography with increases in the posterior putamen, globus pallidum, cerebellum, and supplementary motor cortex.193 Additional metabolic findings have been reported in other genetic and nongenetic forms of dystonia194,195; however, there are no consistent findings across all forms of dystonia.
There are no consistent brain pathologic findings in patients with idiopathic dystonia. Among the various reviews196–200 of primarily idiopathic dystonia, the most frequently cited lesions are in the basal ganglia, including the putamen, the head of the caudate, and the upper brainstem. Hedreen et al201 proposed that the putamen and the striatopallido-thalamo-cortical circuit appear to be the most likely sites in which to search for the unknown defect in primary dystonia.
In a review of the behavioral and motor consequences of focal lesions of the basal ganglia, Bhatia and Marsden202 found that putaminal and globus pallidus (lentiform nuclei) lesions were frequent causes of dystonia. This observation has been confirmed with more recent publications and our own.203 There are several reports of metabolic disorders, particularly those affecting mitochondrial function, causing dystonic symptoms,204 including patients with Leber’s205 or related phenotype,206 partial cytochrome b deficiency,207 ragged-red fibers with complex III and IV deficiency,208 complex I deficiency,209 or other mitochondrial mutation.210 These findings support the concept that disturbed basal ganglia function, including that caused by molecular defects, may be important in the genesis of the dystonia phenotype.
McNaught et al211 recently reported perinuclear inclusion bodies in the midbrain reticular formation and periaque-ductal gray in four cases of DYT1 dystonia, suggesting that impaired protein handling may be a part of the molecular pathology of idiopathic dystonia, and brainstem nuclei may be important in the clinical manifestations. However, detailed histories were not available for all patients to assess whether there were other factors that may have contributed to the observations.
There is also little known about the precise biochemistry of dystonia. Some patients derive benefit from pharmacotherapy with anticholinergics, benzodiazepines, baclofen, dopamine depletors, or dopamine blocking agents. However, from the few autopsies,212 there is normal choline acetyltransferase in the cortex and striatum. In examples of secondary dystonia,213 there is a marked and consistent elevation of norepinephrine in several regions of brainstem. In our case of primary putamenal degeneration, the substantia nigra pars compacta contained a normal number of neurons but decreased tyrosine hydroxylase immunoreactivity.203
Treatment
Various options are available for treating patients with dystonia.214 Aside from the case of levodopa-responsive dystonia, which responds exquisitely to levodopa/carbidopa therapy,215,216 no drug has emerged as uniformly effective, and the surgical approaches are not uniformly beneficial or without risk. One must choose a treatment strategy keeping the risk to a minimum. To avoid potential harm when searching for an effective therapy, surgical therapies and drugs that may cause irreversible harm should be avoided. Very few systematic drug trials have been conducted in dystonia217,218; much of what we know about pharmacotherapy has resulted from empiric observations.219,220 Indeed, in the past two decades, no new oral agent has emerged as specifically effective for patients with idiopathic dystonia, including the focal dystonias.
As genetic and molecular biologic research advances, there is growing concern about the use of ablative CNS surgery. The primary goal of research is to find out more about the etiologic pathophysiology with the intent to develop specific pharmacotherapeutic intervention. With that goal in mind, ablative brain therapy is generally avoided as one might ablate regions of the brain that may hold important receptors to future interventional pharmacotherapy. However, deep brain stimulation of the pallidum and other basal ganglia targets has emerged as an effective therapy for patients with more generalized or focal dystonia that is resistant to other therapies including local injections of botulinum toxin.221–225
Specific pharmacotherapy directed at the underlying identified biochemical defect is available for only a limited number of symptomatic dystonias.226 The most notable is Wilson disease. For tardive dystonia, the best treatment is avoidance of offending medications when possible, and providing the patient with a list of these medications to avoid.
Physical methods temporize symptoms but, in our clinical experience, help very few. Systemic pharmacotherapy provides little relief of symptoms. However, many patients report mild symptom relief with muscle-relaxing agents and anxiolytics such as baclofen and benzodiazepines. Dedo and Izdebski227 described dramatic relief of symptoms by sectioning the recurrent laryngeal nerve. However, the results were not long lasting, due to either reinnervation or central reorganization. The initial favorable reports were temporized by Aronson and De Santo’s228,229 review of 33 patients treated with surgery. By 3 years, only 36% of patients had some persistent improvement and only one of 33 achieved a persistent normal voice. Adverse effects included breathiness, hoarseness, diplophonia, and falsetto. Of the 64% with failed voices at 3 years, 48% were worse than before surgery. Failures were more common among women (77%) than among men (36%). Since that time, other surgical procedures have been reported with limited success. These include a laryngoplastic procedure where there is either an anterior commissure pushback, widening of the anterior commissure, or a vocal fold medialization (for abductor SD).230,231 Myectomy232 and myoplasty233 of the thyroarytenoid (TA), lateral cricoarytenoid (LCA), and posterior cricoarytenoid (PCA) have also been reported to reduce the breaks, but produce unpredictable results. The denervation/reinnervation procedure234 cuts the adductor branch of the recurrent laryngeal nerve (RLN) bilaterally, which is then anastomosed to the ansa cervicalis bilaterally. An LCA myotomy was also added in some patients. The results to date may be promising, but there are not enough long-term vocal results available.
Long-term treatment focal dystonia, and in particular LD, was unrewarding until the advent of local injections of botulinum toxin type A. We began using botulinum toxin type A for the treatment of focal and segmental dystonias in April 1984.235 Improvement in symptoms of LD with local injections of botulinum toxin type A is remarkable and reviewed elsewhere in this volume and supported by consensus statements. The American Academy of Neurology,236 the American Academy of Otolaryngology,237 the National Institutes of Health Consensus Panel,238 and other organizations107,236–242 have reviewed the clinical usefulness of botulinum toxin therapy; the expert panels found this modality of treatment safe and effective for most focal dystonias.
Patients with more than focal dystonia (segmental, multifocal, generalized) are usually treated with pharmacotherapy. However, many patients have benefited from botulinum toxin type A therapy directed toward one or many discrete regions of the body. Pharmacotherapy is usually initiated with an anticholinergic, benzodiazepine, or baclofen. The choice of drug to initiate usually depends on the age of the patient, prior exposure to medications, and other concurrent medications or medical problems. Drug dosage is low initially, and gradually increased as tolerated. If the medication is of no benefit at a dose necessary to cause adverse effects, then it is gradually tapered and discontinued. If a medication is documented to be helpful, then it can be continued, and the next medication is added. In difficult cases, dopamine depleting (e.g., tetrabenazine243,244) and receptor blocking agents245–247 may be added.
Intrathecal baclofen, which has been approved by the Food and Drug Administration (FDA) for patients with spasticity of spinal and cerebral origin, has been used in a limited number of studies and has helped many patients with dystonia. Patients are considered candidates for intrathecal baclofen after they have failed pharmacotherapy, or other conservative treatment. Because of the complexities of patient management, it is recommended that intrathecal baclofen be administered by a team of specialists, including a neurologist and neurosurgeon, familiar with the treatment program, and equipped to manage side effects.248–252
As noted elsewhere, the effectiveness of DBS for the treatment of drug-induced dyskinesias in PD has revitalized interest in the surgical treatment of dystonia. The globus pallidus internus (GPi) appears to be the optimal target for intractable dystonia.253,254 The most dramatic improvement occurs in patients with primary dystonia and those with the DYT1 gene mutation. A lesser degree of improvement may be seen in patients with secondary dystonia.221,222,225,255,256 DBS of the pallidal nucleus recently received a Humanitarian Use Device designation by the FDA.257
Other Cranial Neurolaryngeal Dystonias
Dystonia may affect the soft palate, pharynx, mandible, and other nearby regions. The principles of classification and therapy are similar to those for laryngeal dystonia. It should be emphasized that, associated with their rarity, we have cared for patients who have been misdiagnosed as having glos-sopharyngeal neuralgia or psychogenic disorders, typically “globus hystericus,” who, on further careful examination, have involuntary muscular contractions of the oropharyngeal, lingual, or mandibular musculature. If dystonia is suspected, then appropriate referral to a team with expertise in laryngeal movement disorders is indicated.
Tremor
The appearance of tremor is that of a rhythmical movement of a part of the body. The appearance implies that the movement has a relatively fixed periodicity and possesses an amplitude and waveform that are to some extent invariable over reasonable amounts of time. If these characteristics do not hold, then movements have an irregular appearance and historically have been classified as different phenomena.258 Tremor has been defined as “a series of involuntary, relatively rhythmic, purposeless, oscillatory movements”259 that have been observed in axial, distal, or proximal musculature, or any combination thereof. Tremors have been classified by their etiology or clinical appearance and are characterized by their frequency, amplitude, distribution over the body, and exacerbating and relieving factors.260 The classifications terms are for the most part descriptive or associative and may describe a purported neuroanatomic etiology, such as rubral, basal ganglia; some other causative factor, such as drug-induced, anxiety, or thyroid tremor; frequency; or provoking position/action, such as action-induced or rest tremor (Table 17.6). All individuals have normal physiologic tremor, which occurs with a frequency of approximately 6 to 12 Hz. Abnormal (pathologic) tremor, on the other hand, has been reported to range from 3 to 7 Hz and is considered a sign of neurologic disorder.261 Rest tremor occurs in relaxed, unsupported limbs, and action tremors occur during muscle contraction. Action tremors have been classified further as postural tremors (when holding a position against gravity), contraction tremor (produced by isometric voluntary contraction independent of gravity, such as making a fist), and kinetic or intention tremor (during goal-directed movement such as touching a finger to the nose). Few of these descriptive classifications precisely identify the cause of the tremor and are confounded by overlap. Nevertheless, the descriptive terms may be used as a context from which to consider therapeutic intervention. Pathologic tremor is frequently observed in the limb, hand, and foot musculature of these patients, and vocal tremor may accompany these diseases as well.83,262 The proposed underlying neural bases for tremor include a central mechanism, as in essential tremor and that associated with parkinsonism and dystonia, or a peripheral mechanism, such as the tremor associated with a polyneuropathy.263 This section discusses predominantly those associated with central causes.
| Action | Kinetic | Postural |
| Cerebellar | Midbrain | Primary writing |
| Drug-induced | Multiple sclerosis | Psychogenic |
| Dystonic | Orthostatic | Reemergent |
| Enhanced physiologic | Palatal myoclonic | Rubral |
| Essential | Parkinsonian | Toxin induced |
| Head | Peripheral neuropathy | Vocal |
| Hyperthyroid | Physiologic | Wing beating |
| Intention | Posttraumatic |
Source: Data from Louis ED. Tremor disorders: identification and treatment. Med Update Psych 1997;2:172–178.
Voice Analysis and Characteristics Accompanying Tremulous Diseases
The involuntary, rhythmic, oscillatory movements that affect the distal musculature in patients with tremulous diseases may also affect the muscles of the speech production mechanism and generate rhythmic alterations in pitch and loudness, called “vocal tremor.” Vocal tremor may result in rapid decreases and increases in loudness and pitch or in complete phonation stoppages. Intelligibility and rate of speech may be decreased. Vocal tremor has been described perceptually as “tremulous voice,”262 “wavy voice,”264 or “tremulous, quavering speech,”265
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


