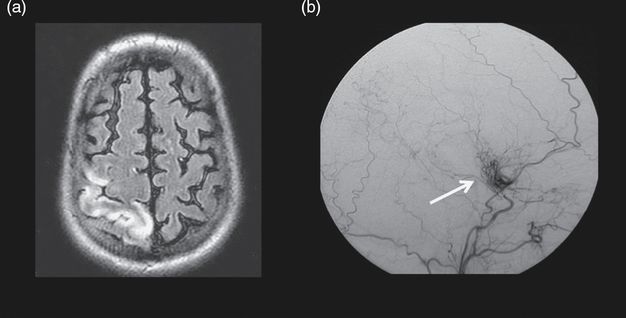
(a) Cerebral MRI showing a recent right cortical infarct on diffusion-weighted images (upper image) and old ischemic lesions in watershed areas on FLAIR MRI (bottom image). (b) Initial (b1, b2, b3) and follow-up (b4, b5, b6) digital subtraction angiography showing right ICA on lateral view (b1, b4), cerebral posterior arteries on frontal view (b2, b5), and branches of external arteries (b3, b6). Moyamoya vessels are seen in the vicinity of the carotid artery apex (black arrow, b1 and white arrow, b4). Note the progression of stenosis on the right ACA (blue arrow, b1), the progression of the leptomeningeal anastomosis arising from PCAs, and the appearance of a transdural collateral (red arrow, b6).
Discussion
The medical history of this patient is highly suggestive of moyamoya disease (MMD). All criteria of MMD are present: (a) the location of steno-occlusive lesions that must involve the terminal portions of ICAs or the proximal areas of ACAs or MCAs; (b) the presence of an abnormal vascular network (of so-called “moyamoya” vessels) observed in the vascular territories close to occlusive or stenotic lesions; (c) the intracranial and bilateral aspect of the arteriopathy; and (d) the exclusion of obvious conditions already known to promote appearance of moyamoya vessels and called moyamoya syndromes [1]. The female sex and Asiatic origin of the patient are additional arguments for MMD as the disease is 10 times more frequent in Japan and Korea than in western countries and affects mostly females (sex ratio around 2:1) [2]. Because of its rarity, data concerning the natural history of MMD, particularly in adults, are still limited and were previously reported only from cohorts of small size. The clinical progression of MMD appears variable; development of severe disability can occur rapidly in some cases but the disease can also remain silent over several decades in other cases. The predictive factors of clinical worsening are not well known. In the present case, MMD was revealed by an acute MCA infarct but remained clinically silent during several years after this event. The patient experienced later hemodynamic transient ischemic attacks (TIAs) as often observed during the course of the disease. Transient or permanent ischemic manifestations can be induced by hyperventilation with crying, exertion, or even after induction of anesthesia for a minor surgical procedure. The presumed mechanism is that normal cortical vessels, already maximally dilated with chronic ischemia, may constrict in response to the decrease of carbon dioxide partial pressure resulting in reduced cerebral perfusion [3]. The severity of cerebral hypoperfusion can be evaluated by measuring the cerebrovascular reactivity during CO2 or acetazolamide administration using SPECT, MR perfusion, or CT perfusion. Decreased perfusion and impaired cerebrovascular reactivity are presumably associated with a higher risk of ischemic manifestations. In the present case, these manifestations were present and led to the decision for revascularization. The efficacy of this type of surgery has not been previously evaluated in a randomized trial. In several reports, the number of ischemic events was found to decrease after revascularization procedures [4–6]. In adults, indications for surgery appear variable between centers and are most often discussed on a case-by-case basis. To evaluate the potential benefit of surgery, the intracranial hemodynamic status has to be evaluated by cerebral perfusion imaging. Detailed analysis of DSA is also needed. In the present case, not only was the vascular reserve impaired despite the development of leptomeningeal and transdural collaterals, but the occurrence of transient ischemic manifestations was also highly suggestive of intracranial hemodynamic failure. In this case, the indirect technique of synangiosis was adopted which consists in using tissues vascularized by the external carotid artery (ECA; spared by the disease) brought into direct contact with hypoperfused cortical areas. Spontaneous development of neovascularization from ECA is supposed to be enhanced by secretion of angiogenic factors from ischemic brain parenchyma. Direct revascularization techniques (such as superficial temporal artery–MCA bypass) are also available for adult patients. When it is successful, the bypass procedure allows a quicker improvement of cerebral hemodynamics. This surgery is, however, associated with a higher risk of hyperperfusion syndrome; the risk can be reduced with specific anesthetic management, which is always needed in MMD to avoid any significant variations in blood pressure and occurrence of hypo- or hypercarbia [7].
Tip
The management of cerebral hemodynamic impairment in moyamoya patients is crucial. In case of ischemic events, the first step is to actively look for triggering factors such as increase of hypotensive drugs, dehydration, or heart failure. The decision of surgery for MMD should follow a multidisciplinary approach for evaluating the spontaneous versus the postoperative risk and be performed by well-trained teams of anesthesiologists and neurosurgeons. For this type of surgery, but also even in the case of minor procedures outside this indication, anesthesia needs particular care to limit the risk of increasing cerebral hypoperfusion.
Case 2. Hereditary moyamoya syndrome
Case description
A 28-year-old man born in Italy with a history of early onset cataract reported repeated episodes of sudden left hemiparesis since the age of 20 years. After the most recent episode, cerebral MRI revealed a recent right-sided cortical MCA infarct and multiple bilateral old subcortical infarcts. DSA showed an occlusion of both ICAs distal to the origin of the ophthalmic artery with a typical aspect of “moyamoya” vessels and leptomeningeal/transdural anastomosis. The posterior cerebral arteries and basilar artery were normal (Figure 14.2). Low dose aspirin was initially recommended. Five years later, the patient had several dyspnea episodes. Echocardiography revealed a severe dilated cardiomyopathy of unknown origin with a significant reduction of the left ventricular ejection fraction (LVEF). Conventional coronary arteriography was normal. From this time, the patient was hospitalized several times for recurrent episodes of heart failure and neurological deficits occurring only when standing and occasionally associated with seizures. Transcranial Doppler suggested hemodynamic impairment on both MCAs. Brain surgical revascularization was declined because of his severe cardiomyopathy. The patient presented with a sudden episode of transient hemiparesis 3 days after his diuretic medication had been increased. Cerebral MRI revealed several new brain infarctions. The patient was 1.50 m tall, had a history of early onset cataract, and had a dysmorphism with bilateral ptosis and long philtrum. Endocrine evaluation revealed partial growth hormone deficiency and hypergonadotropic hypogonadism. Testicular volume was decreased and spermogram showed azoospermia. Information about family history was collected. One of his brothers and the son of his sister had a history of stroke during infancy. A Xq28 deletion removing the BRCC3 and MTCP1 genes was identified in the patient, his brother, and his nephew.
Discussion
This patient has a moyamoya syndrome related to the loss of BRCC3 and MTCP1 genes. Moyamoya syndrome refers to a moyamoya angiopathy associated with other neurological and/or extracranial symptoms, or due to a well-identified acquired or inherited cause. In contrast with MMD, in moyamoya syndrome, the steno-occlusive lesions are not always limited to bifurcations of ICAs. The posterior circulation can be involved and other vascular abnormalities including ectasia, aneurysms, fistula, or mega-dolicho arteries can be observed [8]. In this patient, the characteristics of the intracranial angiopathy are similar to those of MMD. Nevertheless, investigations to identify an acquired condition or an inherited cause of MMS showed several abnormalities. Well-known genetic disorders such as neurofibromatosis type 1, sickle cell disease, or Down syndrome can be easily diagnosed by the presence of café-au-lait macules, sickle cell crisis, or a typical dysmorphism, respectively [9]. But there are several other chromosomal disorders and Mendelian diseases that can lead to a moyamoya syndrome with highly variable penetrance. Questioning about the family history is thus crucial for detecting any abnormality in other affected members and to identify the precise pattern of inheritance. In the present case, all affected individuals were male and related through a maternal lineage which suggested an X-linked pattern of inheritance. Other information may also be crucial such as consanguinity suggestive of an autosomal recessive pattern of inheritance. In this patient, short stature, dysmorphism, dilated cardiomyopathy, azoospermia, and early onset cataract were the main associated features. Investigations must specially include echocardiography, imaging of the aorta and its branches, eye examination, and full body scan. Causative genes have been identified in numerous hereditary moyamoya syndromes. In this family, the disease is caused by an Xq28 deletion that leads to a complete loss of expression of BRCC3 and MTCP1 genes [10,11]. BRCC3 encodes for a member of two protein complexes: a nuclear DNA repair complex and a cytoplasmic complex that might have a role in cardiomyocyte protection. Experimental studies also suggest that BRCC3 plays an important role in angiogenesis. The exact role of MTCP1 is undetermined to date. Finally, the pathophysiology of the disease remains unknown. Genetic testing is important to confirm the diagnosis when it is suspected and allows genetic counselling, especially for females at risk of being asymptomatic carriers and transmitters.
Tip
This case points out the importance of obtaining detailed family information in the presence of moyamoya and of assessing all potentially associated clinical manifestations in order to identify a genetic form of moyamoya syndrome. The diagnosis of some moyamoya syndromes can now benefit from genetic testing and allows specific management with multisystemic follow-up and genetic counseling in family members.
Case 3. Pediatric moyamoya disease
Case description
An 11-year-old girl was first investigated after the occurrence of recurrent episodes of severe headache associated with neurological manifestations since the age of 8 years. Each time, she complained of one-sided paresthesia for 1–2 minutes, followed by ipsilateral hemiparesis and facial palsy lasting 3–5 minutes and sometimes associated with speech disturbances. One or two episodes occurred monthly with complete recovery. The patient had normal cognitive development and no school difficulty was reported. The frequency of these episodes progressively increased and repeated episodes of headache also occurred from age of 10 years. Neurological deficits and headache were not associated. The intensity and frequency of headaches dramatically increased within previous few months. Her clinical examination showed only a slight motor deficit of the left hand. The patient had normal cognitive abilities and no difficulty in practicing sports. Brain MRI and angio-MR revealed features suggestive of moyamoya disease with bilateral ICA stenosis associated with MCA occlusion, initial ACA stenosis, and typical “moyamoya” aspects of the basal collateral network. Hyperintensities related to low flow vessels were detected on MRI. In the right watershed posterior region, an old infarction was detected. A single infarction was also observed in the left deep MCA territory (Figure 14.3). The recurrent neurological episodes were considered as possible manifestations of transient ischemia. Bilateral indirect revascularization surgery was proposed based on the frequency of these TIAs, previous occurrence of two cerebral infarctions on MRI, and obvious hypoperfusion on perfusion-weighted MR-sequences. Although the transient neurological manifestations disappeared after surgery, occurrence of headache did not, although their intensity decreased. Cognitive development remained excellent and no further stroke event was observed. Perfusion assessment 3 months after surgery showed a significant improvement compared to the previous examination (Figure 14.4).
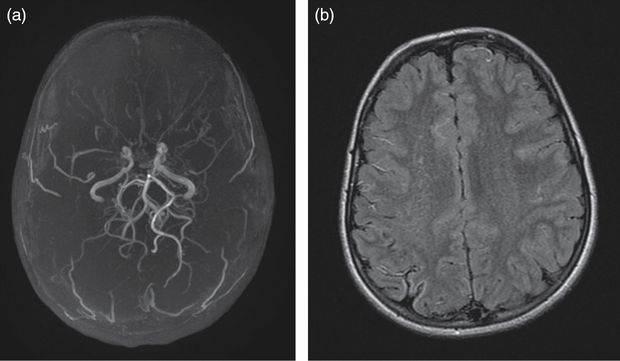
(a) 3D time-of-flight angio-MR showing bilateral terminal ICA narrowing, MCA occlusion, proximal ACA stenosis, and typical moyamoya vessels in the collateral network. (b) Fluid attenuation inversion recovery (FLAIR) brain MRI showing a right-hemisphere small old ischemic lesion and multiple bilateral vascular hyperintensities.
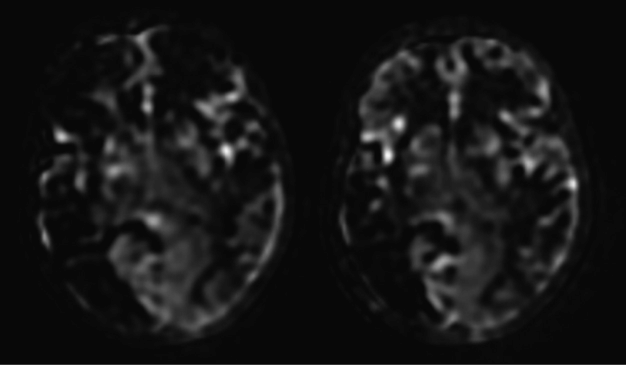
Perfusion MRI with arterial spin labeling showing on the right the preoperative assessment with hypoperfused right MCA and ACA and left ACA territories and on the left the postoperative assessment performed 3 months after bilateral multiple burr hole surgery showing perfusion improvement in the right frontal area.
Discussion
Moyamoya vasculopathy can present with chronic manifestations in children and is not always responsible for stroke events [12,13]. Recurrent episodes of headache as reported in this case are frequent in children with MMD. Repeated episodes of sensory, motor, and phasic deficits were initially thought to correspond to episodes of migraine with aura although the revised International Headache Society criteria for pediatric migraine with aura were not satisfied in the present case. The patient had transient neurological manifestations without headache. In addition, her neurological examination was abnormal. These findings should have prompted brain imaging much earlier in the present case. In pediatric MMD, stroke can occur without overt clinical manifestations and accurate clinical examination can be particularly helpful for diagnosis. A decision for revascularization surgery should be based on multiparametric aspects [314–16]. The occurrence of (recurrent) ischemic manifestations was a strong argument in favor of surgery in the present case as was the presence of old infarcts on MRI. One could object that no actual clinical worsening had been detected over the past 3 years. The recent occurrence of severe episodes of headache was also considered in this case as suggestive of progression of the vasculopathy. Indirect revascularization surgery was considered as the easiest intervention in this patient, who did not present with a rapid progression of the disease that would need an immediate revascularization. Direct artery–artery connection is more difficult with small diameter arteries.
Tip
Pediatric moyamoya may present with recurrent transient ischemic manifestations and headache episodes. Stroke may also occur without obvious clinical manifestations in children. Revascularization surgery may be indicated even in the absence of overt recurrent stroke in the presence of TIAs and silent ischemic lesions on cerebral imaging.
Case 4. Pediatric moyamoya syndrome related to sickle cell disease
Case description
A 5-year-old boy with sickle cell anemia (hemoglobin SS phenotype) presented with abnormal velocities in his annual transcranial Doppler (TCD) evaluation, with time-averaged mean velocity of 221 cm/s for the right MCA, 173 cm/s for the left MCA. His clinical examination and cognitive development were normal. Brain MRI showed bilateral white matter ischemic lesions in anterior watershed regions and bilateral mild MCA stenosis (Figure 14.5). A monthly transfusion program was started. TCD did not return to normal after repeated transfusions over 6 months (228 cm/s for the right MCA and 234 cm/s for the left MCA) and was not even improved after hydroxycarbamide treatment was added. Four years later, transcranial Doppler velocities dramatically dropped in the right MCA, with no measurable flow. In the left MCA, velocities were still abnormal (208 cm/s). Brain MRI and angio-MR showed right MCA occlusion and a typical moyamoya collateral network (Figure 14.6). No further ischemic lesion was detected. Perfusion assessment showed right hypoperfusion, without major left hypoperfusion. Slight school difficulties were noted and adapted education was needed. The boy had recurrent episodes of headache. Unilateral indirect revascularization surgery was decided on using multicraniostomy. Follow-up showed progressive reperfusion in the right hemisphere. Close follow-up with TCD and angio-MR on the left side showed progressive stenosis of the left ICA and progression towards contralateral moyamoya syndrome. Contralateral surgery is now under consideration.
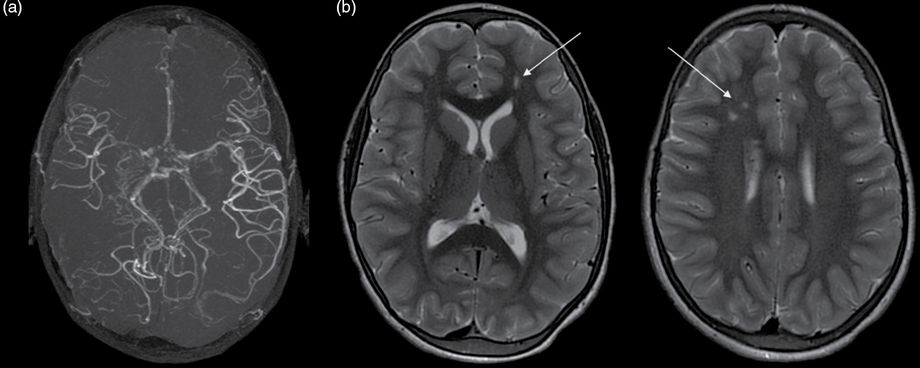
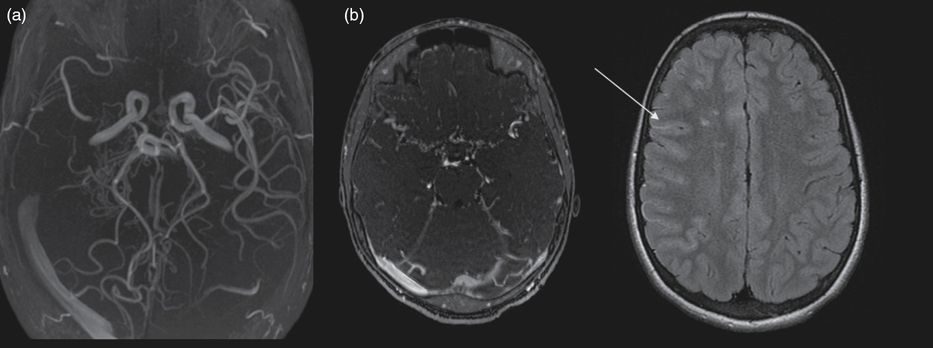
Brain MRI and angio-MR at 9 years of age showing (a) right MCA occlusion, moyamoya collateral network and (b) on FLAIR axial images bilateral ischemic white matter lesions in the anterior watershed areas and, as in Figure 14.1, ivy sign or flow hypersignals: arrow.
Discussion
Cerebral vasculopathy associated with sickle cell anemia (SCA) is frequent and occurs mainly during the first decade [17]. Up to 40% of SCA children with hemoglobin SS or Sβ° will have abnormal TCD screening or MRI markers of cerebral vasculopathy, such as silent infarcts, by the age of 14. Annual TCD screening is recommended from the age of 2 years. Abnormal results (i.e., velocities ≥200 cm/s) lead to chronic transfusion program initiation [18,19]. The chronic reduction of sickle hemoglobin (HbS <30%) dramatically reduces the risk of stroke occurrence. Nevertheless, SCA-associated cerebral vasculopathy is a chronic and progressive condition. In some cases, the transfusion program does not prevent the progression of the cerebral vasculopathy. It is therefore crucial to maintain close clinical and imaging neurological follow-up in children, even under transfusion therapy. This patient had a severe vasculopathy as TCD did not return to normal under transfusion therapy. Hydroxycarbamide, a molecule with promising properties in sickle cell disease and acting by enhancing the fetal protective hemoglobin production, was proposed as it may represent an add-on treatment choice. In this patient, combined transfusion and hydroxycarbamide therapy did not prevent the evolution towards a severe moyamoya syndrome. Although chronic cerebral vasculopathy is frequent in SCA patients, the progression into moyamoya syndrome in well-treated patients is infrequent [20]. Nevertheless, sickle cell disease remains a classical underlying condition that can lead to a moyamoya syndrome. This moyamoya syndrome can initially develop unilaterally and progress later bilaterally. Surgery decision relies on clinical and imaging data used to confirm a typical moyamoya pattern and to assess the clinical, structural, and functional consequences of the vasculopathy (cognitive deterioration, stroke lesions, silent infarcts, and hallmarks of uni- or bilateral hypoperfusion) [18– 20]. Even in the absence of overt stroke, revascularization surgery was decided on in this case due to the progression of the vasculopathy, cognitive deterioration, and recurrent headaches despite maximal medical treatment. Usually, indirect revascularization surgery is targeted in territories showing hypoperfusion as hypoperfusion can presumably trigger development of transdural neovascularization through multiple burr holes. A second contralateral surgery may be needed, as in the present case, if contralateral hypoperfusion is further demonstrated.
Related posts:
 Recognizing cerebrovascular etiologies of “thunderclap†headaches
Choosing the right patients for carotid artery procedural interventions
Transient ischemic attacks (TIAs) – an underrecognized and undertreated disorder
The diagnosis and overdiagnosis of cerebral vasculitis
Dilemmas in endovascular stroke therapy
Underdiagnosis of reversible cerebral vasoconstriction syndromes
Recognizing cerebrovascular etiologies of “thunderclap†headaches
Choosing the right patients for carotid artery procedural interventions
Transient ischemic attacks (TIAs) – an underrecognized and undertreated disorder
The diagnosis and overdiagnosis of cerebral vasculitis
Dilemmas in endovascular stroke therapy
Underdiagnosis of reversible cerebral vasoconstriction syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


