INTRODUCTION
Weakness is one of the most common complaints in all of neurology; hence, both the scope of the term and its differential diagnosis is broad. A patient may have weakness due to a lesion anywhere in the central or peripheral nervous system from the motor cortex to the muscle itself. Often, patients will use the term weakness to describe various functionally limiting symptoms unrelated to loss of strength as with the bradykinesia seen in parkinsonism or the clumsiness experienced with cerebellar ataxia. The concept of weakness is particularly challenging to differentiate from an overall sense of fatigue or asthenia, which is a common complaint in patients with psychiatric (i.e., depression) or systemic diseases such as cancer. It is not at all uncommon for patients to present to the neurologist with a feeling of poorly described generalized weakness, which can be attributed to various nonneurologic or a specific neurologic cause. The charge of the neurologist in this situation is to focus on the backbone of our specialty: Obtain a comprehensive history and perform a detailed neurologic examination to determine if the patient’s complaints are caused by a problem within the nervous system.
MANAGEMENT STRATEGY
Asking the patient about the specific functional consequences of his or her weakness (i.e., trouble climbing stairs, buttoning buttons, opening jars) may help distinguish generalized weakness due to fatigue from weakness with an underlying localizable neurologic cause. Both the history and examination should help the clinician localize the problem to a specific region of the central or peripheral nervous system and give clues to possible etiologies. Symptoms concurrent and associated with the weakness are helpful in formulating a diagnostic impression. Complaints of muscle pain, cramping, or stiffness may invoke a myopathy, whereas numbness or tingling suggests a peripheral neuropathy or central nervous system lesion.
Specific “red flags” in the history or examination may provide important clues and prompt one to institute more acute treatment (
Table 13.1). Once a comprehensive differential diagnosis is made, testing is done to confirm or exclude a specific disorder and may include but not be limited to serum studies, imaging, and electrodiagnostic testing. Many weak patients are encountered in the office setting; however, acute weakness may present in the hospital as well and often warrants a more prompt evaluation and rapid institution of therapy (
Table 13.2).
NEUROANATOMY AND LOCALIZATION OF WEAKNESS
Weakness can be localized to various regions within the neuraxis, from the cortex to the muscle itself, and can be thought of as originating within either the central or peripheral nervous system.
The central, or pyramidal, motor system is composed of the motor cortex, corticospinal tract, and the various other tracts that modify and interact with it (i.e., the rubrospinal and tectospinal tracts). Weakness related to a central process affecting the corticospinal tract or pyramidal system is typically thought to produce a clinical syndrome of upper motor neuron weakness manifested by spastic tone, hyperreflexia, and pathologic reflexes (i.e., Babinski response).
Weakness due to pathology in the peripheral nervous system characteristically produces a pattern of peripheral lower motor neuron weakness consisting of hyporeflexia, muscle atrophy and wasting, reduced tone, and in some cases, spontaneous muscle activity such as fasciculations. The peripheral motor system is typically considered to include the motor neuron within the anterior horn of the spinal cord (or brain stem for cranial nerves), the peripheral nerve, neuromuscular junction, and muscle distal to it. Both the history and neurologic exam will help localize the problem to a particular part of the neuraxis and thus guide one’s differential diagnosis and treatment plan.
CENTRAL (UPPER MOTOR NEURON) WEAKNESS
The corticospinal tract or pyramidal system can be damaged anywhere along its tract from the cortex through the corona radiata to the posterior limb of the internal capsule and down to the brain stem via the cerebral peduncles of the midbrain to the medulla and spinal cord. The highest possible lesion is in the primary motor cortex of the brain, where the largest of the pyramidal cells (Betz cells) lie in layer 5 of the cortex. Responsibility for planning and preparation of movement lies within the supplementary and premotor cortices, which regulate the motor action. The actions of the corticospinal tract are further modulated by the cerebellum as well as the basal ganglia and its tracts, which is also known as the extrapyramidal system. Pathology within this system can lead to either a paucity or exaggeration of movement, and the functional impairment experienced by the patient can be frequently described as weakness.
The neurologic examination and history can point toward a diagnosis localized within either the cerebellar or extrapyramidal system (such as Parkinson disease) and can include such findings as ataxia, bradykinesia (paucity and slowness of movement), tremor, and postural instability. In many cases, the key to localization is “pattern-matching”—actively looking for a pattern or constellation of findings that point to a specific cause of weakness (
Table 13.3).
Cortical Lesions
A variety of diseases can affect the corticospinal tract from large strokes or hemorrhages to tumors and infections. Cortical lesions cause a characteristic pattern of weakness, where one limb is typically preferentially affected due to the distribution of motor representation in the homunculus of the primary motor cortex. For instance, infarcts affecting the middle cerebral artery territory will preferentially affect the arm and face more so than the leg. Often, there are other associated cortical signs such as aphasia or neglect, and patients may also present with headaches or seizures. Cortical weakness can also result from etiologies such as atypical migraine or postictal Todd paralysis; these diagnoses should be considered in the appropriate clinical setting.
Subcortical Lesions
Subcortical lesions, such as those in the internal capsule, will present with more complete unilateral weakness that may or may not spare the face. Demyelinating diseases such as multiple sclerosis (MS) are often implicated in subcortical weakness, although various other etiologies such as infarct or tumor are possible as well. Weakness attributed to a brain stem lesion can present with unilateral weakness of an arm and leg, and the face may be involved as well. There are usually associated symptoms attributable to cranial nerve dysfunction such as diplopia and facial numbness or hemiataxia due to involvement of cerebellar tracts within the brain stem.
Spinal Cord Lesions
After decussating in the pyramids of the medulla, the lateral corticospinal tract is formed within the spinal cord where it travels ipsilateral to the innervated side of the body, lying anterior to the posterior columns and medial to the posterior spinocerebellar tract. Of note, about 10% of fibers in the corticospinal tract do not decussate and travel down into the cord to form the anterior or ventral corticospinal tract. Patients with weakness related to spinal cord pathology will typically present with both motor and sensory complaints, which can be bilateral or localized to one limb. Pain may be present or absent, and its description will vary depending on the cause of weakness. For instance, it may be described as stiffness related to myelopathy, Lhermitte sign (shock-like sensations with back flexion), or diffuse back pain. There may be disturbances in bowel or bladder function requiring prompt evaluation for cord compression.
PERIPHERAL (LOWER MOTOR NEURON) WEAKNESS
Upper motor neurons that form the corticospinal tracts synapse with both interneurons and lower motor neurons within the anterior horn of the spinal cord. The motor unit is defined as one alpha motor neuron within the anterior horn (or brain stem when related to cranial nerves) and all of the muscle fibers it innervates. Pathology within the anterior horn itself may present with a combination of both upper and lower motor neuron signs as seen in amyotrophic lateral sclerosis (ALS) and often includes concomitant bulbar weakness. Patients with weakness localized to the anterior horn will often present with asymmetric weakness, which begins in the bulbar region or within one limb and then progresses segmentally over time. Pain related to muscle cramps and stiffness is a common complaint.
The lower motor neuron exits the spinal cord to form the ventral motor portion of the spinal nerve root, after which it meets the dorsal sensory nerve root past the dorsal root ganglion to form the peripheral nerve. At this location, the dorsal nerve root is commonly susceptible to compression by vertebral disk herniation, and patients may present with radiating pain or sensory loss corresponding to a particular dermatome. A myotome is defined as the group of muscles innervated by one single nerve root. Most muscles are supplied by two or more nerve roots and thus, a single root lesion rarely causes marked weakness due to this overlap within myotomes.
Brachial Plexus
Prior to separating into specific peripheral nerves, the motor and sensory fibers together form a network called a
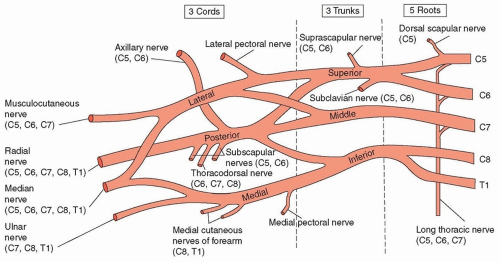
plexus, of which there are two (brachial and lumbosacral). The brachial plexus consists of nerve roots from C5 to T1, whereas the lumbosacral plexus consists of those from L1 to S3. The brachial plexus is composed of three trunks (upper, middle, lower) dividing into two divisions (anterior and posterior), which then form three cords (medial, lateral, posterior) ultimately subdividing and ending in terminal peripheral nerves (
Fig. 13.1). Different peripheral nerves come off the brachial plexus at different levels; some, such as the phrenic nerve, exit immediately off the nerve roots, whereas others form the terminal branches of the plexus. These terminal nerves may be pure motor, pure sensory, or mixed.
Lumbosacral Plexus
The lumbosacral plexus is arranged somewhat differently, with various nerve roots coming together to form large nerves that then continue to differentiate into terminal branches or peripheral nerves. For instance, nerve roots from L4 to S3 come together to form the sciatic nerve, which later then subdivides into the fibular and tibial nerves. Weakness localized to the plexus typically presents with acute or subacute pain in one limb and is most commonly traumatic but can also result from other various etiologies such as infectious, structural, and inflammatory causes. Weakness is localized to one limb, often in the distribution of various nerves. There may be associated sensory complaints but the sensory examination is frequently normal.
Peripheral Nerves
Terminal or peripheral nerves may be affected in isolation (mononeuropathy) or together in a uniform or multifocal pattern. Mononeuropathies are most commonly due to compression, and symptoms and exam findings are restricted to the distribution of one nerve distal to the injury. Polyneuropathies have a broad differential and can be subdivided into those caused by demyelination and those with predominant axonal involvement.
Demyelinating neuropathies typically also involve the nerve roots and can be referred to as radiculoneuropathies. They tend to have a motor greater than sensory predominance, present with more generalized or multifocal rather than length-dependent weakness, and areflexia is typical on examination. Cranial nerves may be affected as well.
Axonal polyneuropathies are usually length-dependent, affect sensory greater than motor function, and spare cranial nerves. The differential for such a polyneuropathy is vast and includes metabolic, infectious, and systemic causes among others. Diabetic neuropathy is by far the most common etiology of length-dependent axonal polyneuropathy.
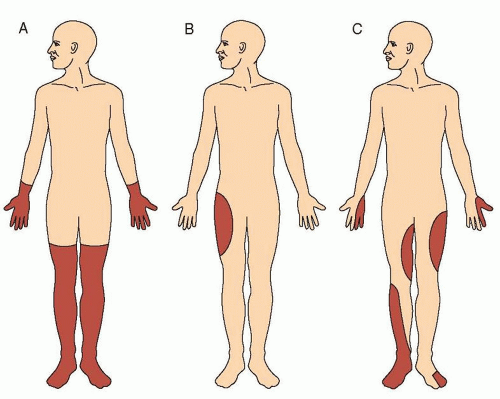
At times, different peripheral nerves can be affected in an asymmetric and patchy fashion (
Fig. 13.2). Such a syndrome of multiple mononeuropathies (also frequently referred to as
mononeuritis multiplex ) is often the result of ischemic injury to different peripheral nerves and present with acute and painful weakness and sensory loss. In these instances, it is crucial to rule out an underlying vasculitis, as management must be prompt to avoid further irreversible injury.
Neuromuscular Junction
Each peripheral nerve ultimately terminates at the motor endplate of the muscle it supplies, where it forms the neuromuscular
junction. At this point, calcium influx into the terminal portion of the neuron leads to binding of acetylcholine-containing vesicles to the neuronal membrane and release of Ach into the postsynaptic space. The Ach-release site is known as the
active zone and is the location of both calcium influx via voltage-gated calcium channels and acetylcholine release. Binding of acetylcholine to its corresponding postsynaptic receptors at the motor endplate leads to opening of sodium channels and consequent depolarization of the muscle membrane and an endplate potential (EPP). The summation of several EPPs is ultimately what leads to a discrete muscle fiber potential and subsequent muscle contraction.
Weakness localized to the neuromuscular junction (see
Chapter 89) often presents with proximal and bulbar weakness, which fluctuates with activity and time of day. Autoantibodies may be directed toward components of the presynaptic membrane (i.e., toward the voltage-gated calcium channel in Lambert-Eaton myasthenic syndrome) or the postsynaptic membrane (i.e., myasthenia gravis, where antibodies are formed against acetylcholine receptors). The muscle fiber action potential causes depolarization of the internal portion of the muscle fiber and leads to activation of calcium channels in the sarcoplasmic reticulum of the myocyte leading to calcium release. Calcium then binds to troponin C on the actin filaments of the myofibril, thereby unbinding tropomyosin from the filament, allowing the binding of myosin. The binding of adenosine triphosphate (ATP) to myosin then allows unbinding and release of actin, thereby leading to muscle relaxation.
Myopathy
Muscle weakness due to pathology within the muscle itself is frequently proximal, symmetric, and progressive. There can be associated facial weakness and weakness of eye movements (such as in mitochondrial myopathies). Myalgias are common, and the sensory examination should be normal. Etiologies are variable and can include inflammatory, congenital, and metabolic causes.
FOCUSED HISTORY DEMOGRAPHICS AND PAST MEDICAL HISTORY
Knowledge of the patient’s age and prior medical history is crucial, as various etiologies are more or less likely depending on the age of the patient. A left hemiparesis presenting in a young woman is more likely to be attributable to an MS flare than a stroke, and the opposite is true in an 87-year-old man with hypertension.
One must be careful to obtain a complete list of medications the patient is taking, as certain exposures may provide diagnostic clues (i.e., statin myopathy). The social history is important to obtain, particularly with respect to toxic exposures and travel, as both toxic ingestion (i.e., alcohol, lead) and various infectious agents may lead to weakness of either central or peripheral origin. A detailed family history is required in cases of suspected hereditary disorders and a pedigree is often helpful. Thus, the age and complete medical, social, and family history of a patient will help formulate an appropriate differential diagnosis based on likelihood and help dictate management.
TEMPO OF WEAKNESS
One of the key questions one must ask is the time of onset and whether the weakness is acute, subacute, or chronically progressive (see
Table 13.3). Focal weakness that begins abruptly (i.e., over seconds to minutes) is most concerning for a vascular event such as a hemorrhage or infarct and these patients must be emergently assessed and evaluated. Subacute onset of weakness has a broader differential and may be due to demyelinating disease such as MS, a mass lesion, or an acute demyelinating neuropathy, among others. Weakness that evolves chronically over months to years is suggestive of a neurodegenerative disease such as motor neuron disease, peripheral neuropathy, myopathy, or a slowly progressive structural lesion.
The tempo of the course of symptoms dictates the urgency with which the patient should be evaluated and managed.
VARIABILITY OF WEAKNESS
Another crucial component of a detailed neurologic history includes asking about the nature of the weakness and whether it is transient, fluctuating, or permanent. Transient weakness that fluctuates with the time of day or with exertion may point toward a neuromuscular junction disorder, MS flare, or a channelopathy (i.e., periodic paralysis).
DISTRIBUTION OF WEAKNESS
The distribution and symmetry of weakness is equally important to characterize when formulating a plan for localization and likely etiology. Weakness affecting one arm and leg on the same side is referred to as hemiparesis. If the arm and leg are affected on opposite sides, this is known as crossed hemiparesis. If the weakness involves only one limb, this is called monoparesis, whereas weakness of both legs is known as paraparesis. The term plegia in the same context refers to complete paralysis.
Certain patterns of weakness give clues regarding localization. Predominantly upper motor neuron weakness of one limb, with or without facial involvement, suggests a cortical lesion in the central nervous system such as a stroke or MS. Given the distribution of the motor homunculus, incomplete upper motor neuron weakness in one limb suggests a lesion in the cortex, whereas complete hemiplegia involving the face is more likely to be localized to subcortical
structures or the brain stem. If certain muscles within one limb (corresponding to a particular myotome or peripheral nerve) are preferentially affected, this may indicate a problem in the peripheral nervous system such as a radiculopathy or mononeuropathy. Generalized weakness can involve all four limbs and be related to a process within the spinal cord (i.e., cervical myelopathy), peripheral nerve (i.e., polyneuropathy), neuromuscular junction, or muscle.
Further characterizing the pattern as being distal or proximal is also helpful. For instance, generalized weakness that is symmetric and distal is commonly related to a polyneuropathy, whereas proximal weakness is more likely referable to a problem in the neuromuscular junction or muscle.
PAIN
Pain can be encountered in the setting of neuromuscular weakness but must be differentiated from the diffuse myalgias that can be related to an underlying systemic medical illness or psychiatric disease. Often, this distinction is difficult to make, as complaints can be nearly identical. Patients with neuropathy often present with burning pain, which is distal and symmetric, and those with radiculopathy or plexopathy may complain of radiating neck, arm, or shoulder pain. Hand pain is a common manifestation of carpal tunnel syndrome, which may eventually progress to hand weakness and clumsiness. Mononeuritis multiplex due to nerve infarcts in the setting of vasculitis can present with acute pain in a variable distribution. Plexopathy is often heralded by acute to subacute pain in the affected limb prior to weakness or sensory complaints. Weakness in the setting of a slowly progressive brain or spinal cord mass may present associated with headaches or back pain.
STIFFNESS AND CRAMPS
Muscle stiffness is commonly encountered in upper motor neuron lesions causing spasticity and can be seen in both weakness due to cortical, subcortical, and brain stem lesions as well as with myelopathy in spinal cord syndromes. Cramps (a localized muscle spasm or persistent contraction) are more of a lower motor neuron/muscle phenomenon and are encountered frequently in various peripheral syndromes. It is seen as a frequent manifestation of motor neuron disease, where patients may complain of frequent cramps and intermittent debilitating muscle spasms. Those with polyneuropathy may often also have cramping or stiffness that is more constant and less paroxysmal.
Various primary muscle diseases include cramps as a primary symptom, and often, the distinction must be made between cramps and more diffuse myalgias. Myopathies characterized by
myotonia (prolonged muscle contraction after voluntary contraction or percussion) such as myotonic dystrophy and certain channelopathies commonly present with cramps as an initial symptom. Metabolic and certain mitochondrial myopathies often also may present with intermittent cramps.
Stiff person syndrome (see
Chapter 94) is an autoimmune disorder characterized by painful muscle spasms caused by diffuse or localized body stiffness usually in the absence of weakness.
VISUAL AND SPEECH DISTURBANCES
If visual complaints are present, it is necessary to characterize them well. A visual field cut or gaze preference may point to a cortical problem, whereas ptosis or diplopia could be a manifestation of a neuromuscular junction disorder or a myopathy.
Trouble with speech in the form of dysarthria must first be differentiated from a language disturbance. The presence of dysarthria may point toward bulbar weakness of various etiologies. A cortical, subcortical, or brain stem lesion may affect tongue fibers; motor neuron disease may present with or lead to bulbar involvement; and similarly, a neuromuscular junction or muscle disorder may involve trouble with speech and swallowing. A complaint of dysarthria should always prompt one to inquire about swallowing difficulties, as this may have important management and prognostic implications.
AUTONOMIC DISTURBANCES
Urinary complaints (most often incontinence or frequency) are most commonly seen with upper motor neuron lesions, such as those involving the paracentral frontal lobe or spinal cord. A complaint of dark-colored urine may indicate the presence of myoglobinuria indicative of muscle breakdown suggesting a primary muscle disease such as McArdle disease. One should ask about autonomic involvement as well (i.e., symptoms of orthostasis, dry mouth or eyes), as this may play an important role in certain neuropathies. Finally, symptoms suggesting cardiac involvement such as syncope, chest pain, and shortness of breath with activity may be worrisome for a concurrent cardiomyopathy as can be seen with certain myopathies (i.e., Duchenne or Becker muscular dystrophy).