Nervous System Formation and Malformation
Gary D. Clark
INTRODUCTION
Induced by underlying mesodermal structures, such as the notochord, and inhibited by surrounding ectodermal tissue, the nervous system begins as a thickened layer of poorly differentiated cells in the developing ectoderm of a 14-day human embryo. By 21 days, ridge-like structures have formed in the lateral most aspects of the neuroectoderm and begin to appose and to close in multiple closure sites, with the first in the cervical region. This forms a tube-like structure termed the neural tube, and this closure is normally complete by 26 days before most women know that they are pregnant. Errors in this process lead to myelomeningoceles (L5 and more rostral) and encephaloceles.
At the most caudal end of this tube is a mixed cell mass of ectoderm and mesoderm termed the caudal cell mass, which is induced by the presence of the neural tube to form the sacrum, the filum terminale, the conus medullaris, and the cauda equina. Errors in these processes lead to tethered spinal cords, caudal regression, fatty filum terminale, sacral pits and tracks, and other sacral defects.
The neural crest cells form from the extreme lateral aspects of the neuroectoderm and give rise to many of the sensory ganglia of cranial and spinal nerves in addition to the sympathetic chain and other structures.
Once the neural tube is completely formed, patterning or segmentation of this tube leads to the beginning of the well-described vesicles of the developing nervous system (prosencephalon, mesencephalon, and rhombencephalon). The prosencephalon divides in the midline to two telencephalic vesicles that will become the cerebral hemispheres. Precursors lining the ventricles of the developing nervous systems become postmitotic and migrate radially to populate developing structures such as the cortex. Axons sprout and cross the midline to form the corpus callosum and thereby connect the two developing hemispheres. Neurons extend axons to connect in a very deliberate and use-dependent fashion to the appropriate targets. This is basic nervous system development. To understand nervous system malformations, some additional genetic, molecular, and basic neurobiology insight is needed.
This chapter will summarize normal and abnormal human central nervous system (CNS) formation and describe the genetic aberrations that cause congenital malformations of the CNS. Much of human CNS development is inferred from animal studies and from abnormal developmental processes. Without being exhaustive, the proteins believed to be involved in the individual components of CNS development and those involved in pathologic alterations of normal development will be listed. The functions of some of the genes responsible for abnormal development can be surmised by examining the pathologic consequences. Hence, one is able to correlate genetic abnormalities with developmental neuropathology. Other genes and molecules involved in abnormal human CNS formation have well-studied counterparts in animals that provide insight into their function in humans.
The study of the genetics of human development is rapidly changing, and important new genetic mechanisms are being described often. Although the information regarding human genes involved in normal and abnormal brain formation was as up-todate as possible at the time that this chapter was written, important new insights into the genetics of human CNS development have likely been made since this was written. Therefore, please visit Online Mendelian Inheritance in Man (OMIM) for more current information (http://www.ncbi.nlm.nih.gov/omim). As is the convention for the time that this chapter is written, terms such as pathogenic variant for those previously termed mutation will be used herein.
Emphasis is placed on important information for the clinician in this chapter, but there is plenty of knowledge of human nervous system development that could be covered herein. However, the space limitations and lack of current applicability to clinical neurology limit the amount of information that can be communicated here. Instead, the covered pathologic entities are presented in the context of the developmental processes that are believed to have gone awry, disorders previously categorized as migration abnormalities might, on the basis of new genetic insights, be found in the discussion of cellular differentiation or segmentation for example. As further insights are made into human brain developmental disorders leading to neurologic disorders, the following categorization scheme will probably have to be altered.
FORMATION OF NEUROECTODERM
The human brain is formed from the neuroectoderm, a placode of cells that are induced by the underlying notochord to differentiate from the ectoderm beginning at 14 days of gestation. The nature of the inducible factor(s) (actually inhibiting factors—bone morphogenic protein 4) involved in this process remain largely unknown. The molecular action of these molecules probably is similar to that of the retinoids that likely bind to a nuclear receptor to promote or suppress specific genes (Fig. 133.1).
NEURAL TUBE CLOSURE
PRIMARY NEURULATION
The neuroectoderm placode develops ridges (folds) laterally and begins to approximate in the region of the future medulla at 22 days of gestation before most women are aware they are pregnant. This closure, a process known as neurulation, results in a tube that continues to extend by the process of approximation of the neural folds in multiple locations rostrally and caudally until a complete neural tube is formed at 28 days of gestation, with the latter period marking the end of caudal neural tube closure (future spinal cord). The rostral neural tube, which closes at approximately 24 days of gestation, serves as the foundation for further brain development; the caudal end of the tube forms the spinal cord.
 FIGURE 133.1 A: Notochord plate induces the neuroepithelium to differentiate from surrounding ectoderm. At the periphery of the neuroepithelium, neural crest forms. B: Neural tube closes. C and D: Rostral neural tube is induced by ventral notochord (Sonic Hedgehog and other factors) to split into two telencephalic vesicles that will become the cerebral hemispheres. (Drawings by Nathan Lycy, http://www.nathanlucy.com/.) |
Although the specific molecules and genes involved in the processes of neural tube closure in humans are largely unknown, clearly a combination of genetic and environmental factors leads to disorders of these processes. Studies in mice suggest that a codeletion of Paxl, a gene for a transcriptional factor that mediates notochord signaling, and Pdgfra, the platelet-derived growth factor alpha gene, may lead to a spina bifida-like phenotype.
Certain ethnic groups experience a higher incidence of neural tube closure defects than do others, and genetic disorders with an apparent autosomal recessive inheritance that include neural tube defects have been described. In addition, teratogens are involved in neural tube pathology. The most notable teratogens for the neurologist are the anticonvulsants valproate and carbamazepine, each of which imposes a risk of 1% to 6% for a neural tube defect in offspring exposed in utero to these common drugs. Whereas folate supplementation appears to prevent neural tube defects in large population studies, this vitamin may or may not be protective when these disorders are associated with the aforementioned anticonvulsants. Given that short-term treatment with folate is probably benign, it seems prudent to offer this vitamin (400 to 4,000 µg daily, depending on risk factors) to women of childbearing age who are on anticonvulsants.
SPECIFIC NEURAL TUBE DEFECTS
In humans, multiple genes and factors probably are involved in the pathogenesis of disorders of neural tube formation and closure. The human disorders of neural tube closure include craniorachischisis (a complete failure of neural tube closure along the entire neuraxis), anencephaly (a failure of anterior neural tube closure), myeloschisis (a failure of posterior neural tube closure), spina bifida (myelomeningocele, a failure of closure of a portion of posterior neural tube), and encephalocele (a partial defect in anterior neural tube closure). Fetal recognition of myelomeningocele with correction in utero prior to 26 weeks’ gestation can dramatically alter the need for subsequent ventriculoperitoneal shunting (reduced by 42%) and reduces the incidence of intellectual disability. The risk is to the mother.
Encephaloceles
Encephaloceles (brain substance outside of the skull) and meningoceles (meninges and cerebrospinal fluid [CSF] only) vary in location, in the amount of brain involved, and therefore in the clinical manifestations of the lesions. In most cases, the neural tube is closed, and the gyral pattern of the protruding brain appears normal. In the western hemisphere, most encephaloceles are occipital
and midline. In the eastern hemisphere, anterior encephaloceles (nasal and frontal) surpasses that of posterior lesions. One should avoid nasogastric tube placement in the situation of a nasal mass in a newborn owing to the possibility of a nasal encephalocele; it is possible in this situation to place the tube in the brain substance.
and midline. In the eastern hemisphere, anterior encephaloceles (nasal and frontal) surpasses that of posterior lesions. One should avoid nasogastric tube placement in the situation of a nasal mass in a newborn owing to the possibility of a nasal encephalocele; it is possible in this situation to place the tube in the brain substance.
Meckel Syndrome
Meckel syndrome, a genetic disorder that involves an occipital encephalocele, cerebellar malformations (molar tooth anomaly— see discussion in Joubert syndrome), microcephaly, renal dysplasia (polycystic kidneys), polydactyly, retinal dystrophy, and other malformations, appears to be caused by pathogenic variants in genes involved in ciliary function. Meckel syndrome is allelic with Joubert syndrome and the same genetic aberrations are present in both disorders. Most of these disorders are inherited in an autosomal recessive fashion, although some are inherited in a dominant fashion and others X-linked dominant or recessive. The genes involved are part of a ciliary complex that determines cell polarity and are important for the migration of early neurons in the posterior fossa.
EMBRYONIC NERVOUS SYSTEM SEGMENTATION
Flexures of the rostral neural tube delineate the primary vesicles of the developing nervous system; these are designated as the hindbrain (rhombencephalon), the midbrain (mesencephalon), and the forebrain (prosencephalon). These primary vesicles then further subdivide into the secondary vesicles that later will form the adult brain structures. The hindbrain consists of the metencephalon and myelencephalon; these structures will become the pons, the cerebellum, and the medulla oblongata of the adult. Rostrally, the mesencephalon will become the midbrain of the adult. The forebrain is further divided into the telencephalon and the diencephalon. The telencephalon will become the cerebral hemispheres, and the diencephalon will become the thalamus and hypothalamus of the adult.
TRANSCRIPTIONAL FACTORS AND HOMEOBOX GENES
Regional specification of the developing nervous system is an important step in human CNS development and is likely under the control of a number of genes that encode transcriptional factors and of a number of molecules that influence these genes. These genes were first described in Drosophila and are involved in the regional specification of the fly embryo. Not surprisingly, the function of the proteins encoded by the comparable genes in mammals differs considerably from that in fruit flies, but the general role of these proteins appears to be that of regional specification of clones of cells destined to form structures in the fully developed nervous system.
Transcriptional factors are proteins with distinct sequences that participate in DNA binding and thus influence DNA. In Drosophila, transcriptional factors encoding homeobox genes, empty spiracles, ems have been shown to specify head structures. For example, the rudimentary structures of mutant flies that are devoid of ems seem to be a result of failure of regional specification of a clone of cells that are destined to form those structures. In humans, pathogenic variants in the comparable gene EMX2 may result in schizencephaly, cleft in the cortical mantle. The clefts in this disorder extend from the pia to the ventricle and are lined with a polymicrogyric gray matter (see the discussion in “Polymicrogyria”). The pia and ependyma are usually in apposition, especially in severe cases. The defect is termed open lipped if the cleft walls are separated by CSF and closed lipped if the walls appose. These clefts may be unilateral or bilateral and the prognosis appears to be dependent on location, bilateral occurrence, or extent of the lesion. Bilateral schizencephaly is associated with mental retardation and spastic cerebral palsy; affected patients often are microcephalic. Seizures almost always accompany severe lesions, especially the openlipped and bilateral schizencephalic clefts. The exact frequency of seizures in patients with the less severe lesions is uncertain, nor is the incidence of asymptomatic schizencephaly. Most patients in whom schizencephaly is diagnosed undergo neuroimaging because of seizure. Therefore, a bias in favor of a universal occurrence of seizure in this disorder is noted. Hence, patients with schizencephaly who do not have epilepsy might exist, but the malformation remains undetected because no imaging is done. With fetal imaging, asymptomatic schizencephaly has been noted (Fig. 133.2).
Seizure type and onset may vary in this disorder. Patients may experience focal or generalized seizures. Some will present with infantile spasms. The onset varies from infancy to the early adult years. Seizures may be easily controlled or may be recalcitrant to standard anticonvulsant therapy.
Improvements in neuroimaging have enhanced the recognition of these disorders and have broadened the spectrum of the radiographic appearance of schizencephalic lesions. The lesions may occur in isolation or may be associated with other anomalies of brain development. An especially common association is made between septo-optic dysplasia and schizencephaly because as many as 50% of patients with septo-optic dysplasia also have schizencephaly.
Other genes for transcriptional factors have been described in the mouse. Pax3, PaxS, Pax6, Dlxl, DIx2, Dbx, and the Hox genes are found in specific brain regions. In general, the Pax genes tend to be expressed in midbrain and the Dlx genes tend to be expressed in the ventral forebrain, ventral telencephalon, and dorsal telencephalon; Dlx2 is important for the genesis of GABAergic interneurons from the median ganglionic eminence. The Hox genes seem to specify the neuromeres and rhombomeres of the hindbrain.
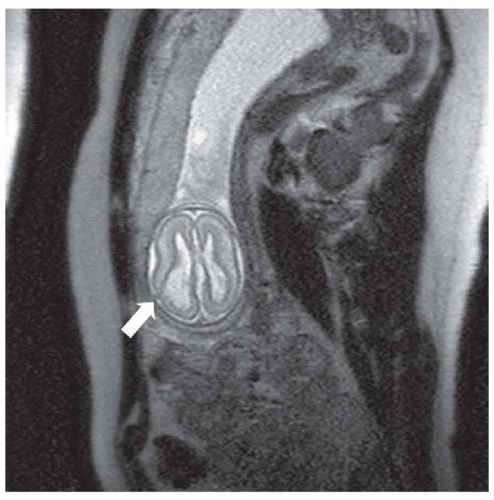 FIGURE 133.2 Segmentation or patterning defect— schizencephalic cleft in a developing fetus at 21 weeks’ gestation. Arrow points to the cleft in the developing cortex. |
Furthermore, in an interesting link to the ventral inductive events described later, when applied to proliferating cells at critical times in development, the protein sonic hedgehog can alter the expression of the homeobox genes. This ties the inductive proteins to the expression of homeobox genes and gives a hint as to the mechanisms involved in inductive processes. Further, a gradient of retinoic acid in a rostral (less) to caudal (more) fashion seems to be important for Hox and other patterning gene expression patterns in the hindbrain, neck, and other structures of the head. Retinoic acid activity also decreases the sonic hedgehog signaling (see “Ventral Induction [Prosencephalon Cleavage]”). It is for this reason that retinoids are highly regulated and should not be given to women who may become pregnant.
SEPTO-OPTIC DYSPLASIA
Septo-optic dysplasia (de Morsier syndrome) is a disorder characterized by absence of the septum pellucidum, optic nerve hypoplasia, agenesis of the corpus callosum, and hypothalamic dysfunction. This disorder should be considered in any patient who exhibits at least two of these, and all such patients should have hypothalamic function screening. Fifty percent of patients with septo-optic dysplasia have schizencephaly. Although this is a rare disorder, genetic syndromes with this phenotype and with some risk of recurrence have been described. For instance, pathogenic variants in homeobox gene expressed in ES cells, HESX1, have caused a dominantly inherited disorder in siblings. This disorder can be suspected in utero but magnetic resonance imaging (MRI) of the fetus may be necessary to assure a proper diagnosis. The prognosis for development is highly variable, with learning, intellectual, and motor impairments often described. Asymptomatic patients probably only come to medical attention if there are needs for neuroimaging. Pathogenic variants in COL11A2 and PAX6 have also been described. Because most of the genes related to this phenotype are undiscovered and no genetic panels exist, it is recommended that clinicians consider exome sequencing in order to delineate the recurrence risk in a family.
VENTRAL INDUCTION (PROSENCEPHALON CLEAVAGE)
NORMAL PROSENCEPHALON DIVISION
The telencephalon is formed by medial division of a rostral single tube-like structure (prosencephalon); the two vesicles (telencephalon) formed in this division become the two cerebral hemispheres. The ventral and anterior portion of this division is induced by midline facial structures and the notochord via soluble factors. Abnormalities of this induction and division lead to midline abnormalities of the brain such as holoprosencephaly. These disruptions of normal development occur before 42 days of gestation.
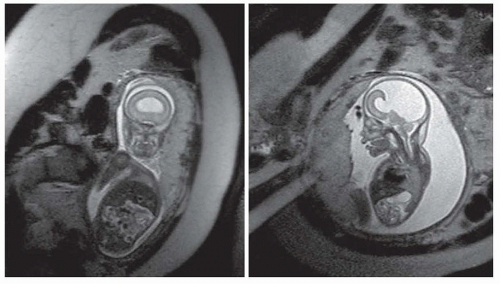 FIGURE 133.3 Failure of telencephalic cleavage— holoprosencephaly in a fetus at 32 weeks’ gestation. Note the single ventricle with no division of the cerebrum into hemispheres on a coronal view (left panel). On a sagittal view (right panel), a dorsal cyst is noted that is typical for the alobar holoprosencephaly. |
Sonic hedgehog, which was described first in Drosophila as a soluble factor (protein) that influences dorsoventral patterning of the developing embryo, is probably the most important of the soluble factors influencing ventral induction. Sonic hedgehog is expressed in the notochord (also in ventral forebrain and floor plate— future facial structures), interacts through a well-defined signaling pathway that includes PTCH, a human homolog of patched, and alters the expression of transcriptional factors (homeobox and other related gene products).
Other molecules of interest in this inductive process are the retinoids (discussed previously in segmentation), which are lipids capable of crossing membranes and have been shown to exist in gradients across embryos. Retinoic acid can alter the pattern of transcriptional factors in neuroepithelial cells, and it can also downregulate sonic hedgehog, perhaps explaining some of the midfacial defects and holoprosencephaly seen in retinoid embryopathy. Also, cholesterol and cholesterol-derived lipids serve as a cofactor for sonic hedgehog, thus perhaps explaining telencephalic cleavage issues (holoprosencephaly) in Smith-Lemli-Opitz syndrome (7-dehydrocholesterol reductase deficiency).
DISORDERS OF VENTRAL INDUCTION: HOLOPROSENCEPHALY
The holoprosencephaly syndromes are heterogeneous disorders of ventral induction and telencephalic cleavage that result from a failure of the prosencephalic vesicle to medially cleave normally. At least three forms of this disorder have been described: alobar, semilobar, and lobar. In the alobar form, the telencephalic vesicle completely fails to divide, producing a single horseshoeshaped ventricle, sometimes with a dorsal cyst, fused thalami, and a malformed cortex (Fig. 133.3). In the semilobar form, the interhemispheric fissure is present posteriorly, but the frontal and, sometimes, parietal lobes continue across the midline. In the lobar form, only minor changes may be seen: The anterior falx is absent, the frontal lobes and horns are hypoplastic, there may be partial fusion of the thalami, and the genu of the corpus callosum may be abnormal. Holoprosencephaly is a common human malformation leading to spontaneous abortion.
Because sonic hedgehog and patched are expressed in the developing face, it is not surprising that holoprosencephaly is associated with a spectrum of midline facial defects. These include cyclopia, in which there is a single central eye and a supraorbital proboscis; ethmocephali, in which the nose is replaced by a proboscis located above the hypoteloric eyes; cebocephaly, in which hypotelorism and a nose with a single nostril are seen; and premaxillary agenesis, with hypotelorism, a flat nose, a single frontal incisor, and a midline cleft lip.
Only children who have the semilobar and lobar forms are known to survive for more than a few months. An infant affected with the severe form is microcephalic (unless aqueduct stenosis and hydrocephalus are present), hypotonic, and visually inattentive. In infants with the less severe forms of holoprosencephaly, myoclonic seizures frequently develop and, if the infant survives, autonomic dysfunction, failure to thrive, psychomotor retardation, and atonic or spastic cerebral palsy often are present. Some infants with the lobar form may be only mildly affected. Pituitary defects may be associated with these malformations and may result in neuroendocrine dysfunction.
Holoprosencephaly has been reported to be associated with maternal diabetes, retinoic acid exposure, cytomegalovirus, and rubella. Chromosomal abnormalities associated with this disorder include trisomies 13 and 18; duplications in 3p, 13q, and 18q; and deletions in 2p, 7q, 13q, and 18q. Autosomal dominant forms exist in which the pathogenic variant is in sonic hedgehog or in ZIC2. In this form, the clinical features vary. In their mildest form, presence of a single central incisor, attention deficit disorder, or a choroid fissure coloboma may be the only clinical manifestation of an autosomal dominant disorder.
NEURONAL AND GLIAL PROLIFERATION
NORMAL CELL PROLIFERATION
Lining the interior of the newly developed telencephalic vesicles is a proliferative, primitive neuroepithelium. Neuroepithelial processes extend from the ventricular surface to the pial surface, and the nuclei of the primitive neuroepithelial cells move from the cortical surface in a premitotic phase to a mitotic phase near the ventricle. Cells divide at the most ventricular aspects of the developing telencephalon and, after division, move back toward the pial surface. The pial processes of neuroepithelial cells near the ventricle often will detach from the cortical surface before a new cycle begins.
Neuroepithelial cells divide in so-called proliferative units such that each unit will undergo a specific number of divisions resulting in the appropriate number of cells for the future cortex. Abnormalities in the number of proliferative units or in the total number of divisions can lead to disorders of brain manifested by abnormal brain size and, therefore, an unusually small (microcephaly) or large head (macrocephaly). Disorders in which too many cells are generated in the proliferative phase result in megalencephaly (large brain) or, if proliferative events go awry on only one side of the developing cortex, hemimegalencephaly. Once the appropriate complement of cells is generated in the proliferative phase, the cells that will become the neurons of the cerebral cortex become postmitotic and are referred to as neuroblasts. Others undergo a programmed cell death—apoptosis. The genetics of apoptosis are best characterized in the simple nematode Caenorhabditis elegans. Approximately 10% of the cells generated during development of this worm will undergo apoptotic or programmed cell death. Pathogenic variants leading to smaller or larger numbers of surviving cells result in smaller or larger nematodes, respectively. The molecular characterization of these pathogenic variants has led to identification of a number of “death” genes and of genes that prevent apoptosis. The mammalian counterparts of the nematode death genes encode enzymes that are cysteine aspartate-specific proteases, also known as caspases. At least one function of such enzymes is that of an interleukin-converting enzyme common in the inflammatory response of the body. These enzymes have been shown to promote neuronal death; in addition, when an animal is produced without caspase 3, a larger than normal brain results.
DISORDERS OF NEURONAL AND GLIAL PROLIFERATION
Microcephaly
Although primary familial microcephaly may be a normal variant, in the classic symptomatic form, clinical and radiologic examinations reveal a receding forehead, flat occiput, early closure of fontanelles, and hair anomalies such as multiple hair whirls and an anterior cowlick. Neuroimaging may show small frontal and occipital lobes, open opercula, and an uncovered cerebellum. The cortex may appear malformed (polymicrogyria, pachygyria, diminished white matter).
Neurologic findings also vary. Only mild psychomotor retardation may be noted, sometimes associated with pyramidal signs, or more severe intellectual disability, seizures, and an atonic cerebral palsy might be present. Primary microcephaly is seen in many genetic syndromes and, in its isolated form, may be autosomal recessive. Autosomal dominant and X-linked transmission has also been reported. Microcephaly vera is the term applied to this genetic form of microcephaly. Affected children present with a head circumference that is usually more than two standard deviations below the mean, hypotonia, and intellectual disability. They later show intellectual disability, dyspraxias, motor incoordination, and sometimes seizures.
Destructive lesions of the forming brain, such as those caused by teratogens and by infectious agents, also may result in microcephaly. Teratogens of note are alcohol, cocaine, and hyperphenylalaninemia (maternal phenylketonuria). Intense radiation exposure (such as in a nuclear explosion) in the first trimester can cause microcephaly. Microcephaly and intracranial calcifications are likely due to well-recognized in utero infections caused by cytomegalovirus, toxoplasmosis, or the HIV, among others.
Although now usually recognized in utero, patients with pathologic microcephaly or pathologic macrocephaly have been, in the past, identified by their pediatricians. Neuroimaging may reveal an etiology in such patients. Because multiple genetic or teratogenic etiologies must be considered, one must use neuroimaging as a clue to possible etiologies (presence of malformations, calcifications, etc.). The prognosis varies greatly and depends on the etiology. Familial microcephaly and familial macrocephaly with benign outcomes have been described.
Megalencephaly and Hemimegalencephaly
The terms megalencephaly and hemimegalencephaly refer to disorders in which the brain volume is greater than normal (not owing to the abnormal storage of material); usually, the enlarged brain is accompanied by macrocephaly or a large head. Although
considered by some to be a migration disorder, the increase in brain size in these disorders appears to be attributable to errors in neuroepithelial proliferation, as the microscopic appearance of the brain is that of an increase in number of cells (both neurons and glia) and in cell size.
considered by some to be a migration disorder, the increase in brain size in these disorders appears to be attributable to errors in neuroepithelial proliferation, as the microscopic appearance of the brain is that of an increase in number of cells (both neurons and glia) and in cell size.
Typically, patients are noted to have large heads at birth and may manifest an accelerated head growth in the first few months of life. Children with megalencephaly or hemimegalencephaly may come to medical attention when presenting with seizures, a developmental disorder (intellectual disability), hemihypertrophy, or a hemiparesis (opposite the affected hemisphere). Seizures vary both in onset and in type and usually are the most problematic symptom, sometimes necessitating hemispherectomy or callosotomy.
Approximately 50% of patients with linear sebaceous nevus syndrome have hemimegalencephaly associated with HRAS or KRAS somatic-mosaic pathogenic variants. Many patients with hypomelanosis of Ito also have hemimegalencephaly. The neuropathologic and clinical pictures in these conditions appear to be identical to the isolated hemimegalencephalies. Interestingly, the genetic aberrations suggest an upregulation of the mammalian target of rapamycin (mTOR) pathway via a downregulation of phosphoinositol kinase 3 (PI3 kinase).
Microscopic examination of the affected brain usually reveals an increase in cellularity, large bizarre neurons, enlarged glia, cortical lamination defects, and heterotopias. The cortex usually is thickened, and malformed neurons are disturbed in polarity. Interestingly, the cytologic basis for the increase in brain size may be the abundant cytoplasm of individual cells.
NEURONAL DIFFERENTIATION
At the time of neuronal differentiation, the neural tube consists of four contiguous layers: (1) the ventricular zone, which gives rise to neurons and all the glia of the CNS; (2) the subventricular zone, which is the more superficial layer and is the staging area from which postmitotic neurons begin to differentiate and to migrate; (3) the intermediate zone, which is the contiguous, more superficial zone and which is destined to become the cortical plate and the future cerebral cortex; and (4) the marginal zone, which is the outermost zone and is composed of the cytoplasmic extensions of ventricular neuroblasts, corticopetal fibers, and the terminal processes of radial glia (which, at this time, are completely spanning the neural tube).
Differentiation of neuroepithelial cells begins in the subventricular layer at approximately gestational day 26. Neuroepithelial cells were destined to become neurons at the time of the final mitotic division of the neuroepithelial cell precursor before moving to the subventricular zone, the staging area for neuronal migration. At this point, these neuroblasts lack electrically polarized membranes as would commonly be seen in neurons. The fate of the neuroblast probably is determined before this final mitosis has occurred, as the postmitotic neuroblast has the properties of many neuron types. The older, larger pyramidal cells are the first cells to be born and probably differentiate early in order to act as targets or barriers in the migration of the nervous system.
No disorders of solely neuronal differentiation have yet been identified, although some disorders may be found to fit into this category. For instance, premature neuroblast differentiation could result in an inability of these cells to migrate and thus could be manifest as a migration abnormality. The megalencephaly and hemimegalencephaly syndromes may yet be found to be the results of disturbances in differentiation, as discussed earlier. Disorders such as tuberous sclerosis, in which both tumor development and areas of migration abnormalities are seen, seem to be a differentiation disorder due to upregulation of mTOR. The brain manifestations of this disorder include hamartomas of the subependymal layer (tubers), areas of cortical migration abnormalities (cortical dysgenesis), and the development of giant cell astrocytomas in upwards of 5% of patients. Two genes for tuberous sclerosis have been identified: TSC1 (encodes for hamartin) has been localized to 9q34 and TSC2 (encodes for tuberin) has been localized to 16p13.3. Both proteins suppress mTOR and when lost or dysfunctional lead to overactivation of this important protein in development. The frequency of gene abnormalities in tuberous sclerosis patients has been estimated to be almost equally distributed between TSC1 and TSC2. Interestingly, recently type IIB cortical dysplasias were found to harbor human papilloma virus 16 and its oncogenic protein E6, which exerts its effect by upregulating mTOR by, in part, depressing TSC2.
NEURONAL MIGRATION
NORMAL MIGRATION
At the most rostral end of the neural tube in the 40-day-old fetus, the first mature neuron arrives at the developing cortical surface. These first neurons are the Cajal-Retzius cells, which are the major neurons of the cortex by day 43. Cajal-Retzius cells, along with corticopetal nerve fibers, form a so-called preplate. These cells will be the major cell type of the most superficial layer of the cerebral cortex, layer I. At the same time that the Cajal-Retzius cells are arriving at the most superficial layer of cortex, other pioneering neurons differentiate and form a so-called subplate. The pioneering neurons of the subplate and the preplate act as the police officers of the developing nervous system and define the limits of the developing cortical plate, which will become the six-layered cortex of the adult brain. Most of the cells of the subplate will die postnatally in a programmed cell death (see earlier discussion of apoptosis).
Near the end of the proliferative phase of neurodevelopment, billions of postmitotic neurons are poised to begin the trip to the cortical surface and to form the cortical plate. This tremendous number of neurons accomplishes this task, for the most part, by attaching to and migrating along radial glia (radially spanning from ventricle to pial surface) in a process known as radial migration (Fig. 133.4). In the process of migration, the deepest layer of the cortical plate forms before the other layers. Therefore, the first neurons to arrive at the future cortical plate are layer VI neurons. More superficial layers of cortex then are formed such that the neurons of layer V migrate and pass the neurons of layer VI; the same process occurs for layers IV, III, and II. The cortex therefore is formed in an inside-out fashion (see Fig. 133.4).
The molecules and the interactions between neurons and glia are extremely important in this process of neuronal migration. Reelin, the protein involved in the migration mouse mutant, reeler, appears to be one such important molecule. Reelin appears to promote an attachment of neurons to glia and, when abnormal, as in the mouse mutant reeler, it leads to an inverted cortex such that the most superficial neurons are the first to arrive and the later-arriving neurons are deeper. This mouse mutant cortex appears to be due to an abnormally adhesive interaction between neurons and glia. Additional molecules that appear to act in an adhesive manner are laminin, astrotactin, L1 antigen, fibronectin, neural cell adhesion molecules (NCAM), and adhesion molecule on glia (AMOG).
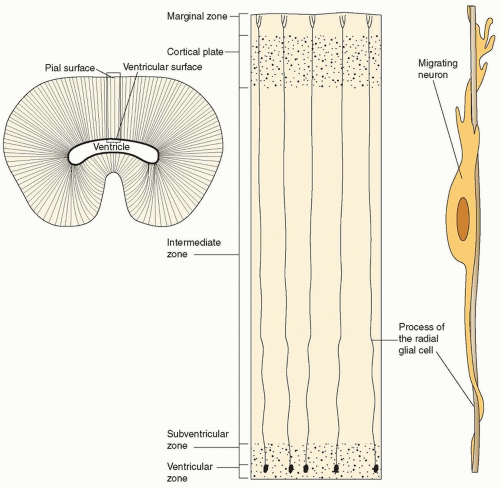 FIGURE 133.4 Neuronal radial migration in cortical development—upper left depicts a single cortical vesicle stained for glia. Glia radially project from the ventricle to the cortical surface. The right figure depicts the apposition of the migrating neuron to a radial glial cell. Cells migrate past their predecessor to deposit in an inside out fashion. |
Neuroblast movement on radial glia involves an extension of a leading process, a neural outgrowth having an orderly arrangement of microtubules. Microtubules are cytoskeletal elements with a polymerizing (positive) end and a depolymerizing (negative) end. They serve as the major structural element that gives shape to long neural processes. When microtubules depolymerize, or slide, long neural processes are shortened. Shortening of the leading process of migrating neurons has been associated with forward movement of the soma of migrating neurons in vitro. Cytoskeletal changes in leading processes have been purported to be responsible for this shortening and somal movement.
A possible mode of movement in neuronal migration on glia would be the attachment of the neuron to a matrix secreted by either the glia or the neurons. This matrix is likely to consist of the aforementioned adhesion molecules. The attachment of the neuron would be through integrin receptors, cytoskeletal-linking membrane-bound recognition sites for adhesion molecules. That attachment serves as a stronghold for the leading process and soma of the migrating neuron. Shortening of the leading process owing to depolymerization or shifts of microtubules results in movement of the soma relative to the attachment points. This theory of movement of neurons also must include a phase of detachment from the matrix at certain integrin receptors so that the neuroblast can navigate successfully along as much as 6 cm of developing cortex (the maximum estimated distance of radial migration of a neuroblast in humans). Finally, the movement of cells must stop at the appropriate location, the boundary between layer I and the forming cortical plate. Therefore, some stop signal must be given in order for the migrating neuroblast to detach from the radial glia and begin to differentiate into a cortical neuron. This is obviously deficient in such disorders as cobblestone lissencephalies (discussed in the following section).
Other forms of neuronal migration occur in brain development. Some evidence exists for a tangential migration of neurons in the cortex and in the early granule cell migration in the cerebellum. A so-called chain migration of neurons on other neurons occurs in the formation of the olfactory bulbs. In this chain migration, neuroblasts from the subventricular zone of the lateral ventricle
migrate to the olfactory bulb through a sheath of glial cells, but the actual migration occurs on other neurons.
migrate to the olfactory bulb through a sheath of glial cells, but the actual migration occurs on other neurons.
CORTICAL NEURONAL MIGRATION DISORDERS
Many clinical entities are associated with neuronal migration disorders. In some, abnormalities are limited to the nervous system but, in others, malformations involving other organs also are present. The responsible genetic mechanism has been identified in some of these disorders, and new genetic mechanisms are being identified regularly. Although the role of the gene product in producing many of these migration disorders may not yet be entirely clear, the disorders provide important clues to mechanisms that are responsible for normal brain development.
Modern neuroimaging techniques, particularly MRI, have allowed the recognition of major migration disorders. Some of these disorders are associated with typical clinical features that might alert the clinician to the presence of such abnormalities even before imaging is obtained. In other disorders, the clinical features are so varied that a strong correlation between imaging and clinical presentation does not exist. High-field MRI and other research techniques may further refine the clinician’s ability to refine diagnoses.
Lissencephaly
Although the term lissencephaly (smooth brain) refers to the external appearance of the cerebral cortex in those disorders in which a neuronal migration aberration leads to improper number of neurons at the surface of the cortex (Fig. 133.5), the most important observation in such cases is the thickened cortex with neurons in what should be white matter. In such deficits of migration, gyri and sulci do not form properly because the cortical-cortical attractive forces that result from strong associations are decreased, owing to improper axon pathways (i.e., the targets for synapses are malpositioned). It is important to note that the term lissencephaly is applied to cortical disorders in which there is a thickened cortex (migration deficit) and a cortical surface abnormality; rarely, there is a completely smooth brain. At least two types of lissencephaly have been identified: type I, or classic, lissencephaly and type II, or cobblestone, lissencephaly. This classification is based on both the external appearance of the brain and the underlying histology.
TYPE I (CLASSIC) LISSENCEPHALY
Type I lissencephaly most often accompanies the Miller-Dieker syndrome. In this disorder, an inadequate radial migration of neurons appears to have taken place. The cortex is described as being composed of a four-layered abnormal sequence consisting of an outer molecular layer (layer 1), which is similar to normal layer I; a disorganized cellular layer of neurons near the normal location for the outer cortex (layer 2) (cells that should be in layers II to VI of the normal cortical plate); a cell-sparse zone (layer 3); and a heterotopic zone of neurons that have been arrested in neuronal migration (layer 4). The most extensive layer in this lissencephalic sequence is layer 4, the heterotopic zone. The neurons and cells of this zone have the appearance of cells that would normally be in layers II to IV of the normal cortical plate; however, these neurons are highly disorganized. Therefore, the later waves of neuronal migration that form the outer cortical plate appear to be those most affected by the migration abnormality in this disorder.
Hallmarks on imaging are a lack of opercularization (covering of the sylvian fissure), large ventricles with colpocephaly (fetallike configuration of the occipital horns), and agyria or pachygyria (see Fig. 133.5). The corpus callosum is almost never absent, and the posterior fossa has a normal appearance on neuroimaging (except in lissencephaly with ambiguous genitalia). Head size typically is in the low-normal range at birth, but most patients develop microcephaly owing to a decreased rate of brain growth over the first year of life. Nearly all patients with this disorder will develop seizures within this first year, and more than 80% of them will have infantile spasms. This seizure frequency is far greater than that seen in other neuronal migration disorders.
In the Miller-Dieker syndrome, facial dysmorphism, cardiac abnormalities (40%), sacral abnormalities (70%), deep palmar creases and, in male patients, genital abnormalities (70%) may be seen. The sacral abnormalities include deep sacral dimples, sacral pits, and sacral tracks. Facial abnormalities include upturned nares; a short nose; a thin, “pouty” upper lip; a long philtrum; micrognathia; and bitemporal hollowing. Although the bitemporal hollowing may be the result of the underlying brain abnormality, the other facial features would be difficult to explain on the basis of the brain abnormality alone. Therefore, these abnormalities are believed to result from deficits of genes near the lissencephaly gene on the 17th chromosome. Larger deletions of the distal short arm of chromosome 17 appear to result in the full Miller-Dieker phenotype, whereas microdeletions of just the lissencephaly gene LIS1 result in isolated lissencephaly. Therefore, a deletion in the lissencephaly gene appears to be sufficient for the brain abnormalities, but other genes (14-3-3) must be deleted for the full phenotypic manifestations of the Miller-Dieker syndrome to be seen.
Miller-Dieker lissencephaly is one of the migration disorders of brain in which the responsible genetic defect has been identified. By both molecular and cytogenetic techniques, deletions in the terminal portion of one arm of chromosome 17 can be found in approximately 90% of Miller-Dieker lissencephaly cases. These patients typically have dysmorphic features and other congenital anomalies (see earlier discussion). The deletions of the terminal part of chromosome 17 in these cases have included microdeletions, ring 17 chromosome, pericentric inversions, and a partial monosomy of 17p13.3. These genetic abnormalities can be the result of an inherited unbalanced translocation from a parent with a balanced translocation involving this region of chromosome 17; multiple mechanisms of inheritance similar to this have been described for this disorder. Of course, a parent with a balanced translocation of chromosome 17p is at a greatly increased risk of having another child with Miller-Dieker lissencephaly. In families that are affected in this manner, screening with amniocentesis can be performed in subsequent pregnancies.
Current chromosomal microarray testing will detect deletions of LIS1 and of the neighboring 14-3-3 gene, thus genetically distinguishing Miller-Dieker syndrome from isolated lissencephaly. Some patients with isolated lissencephaly (no facial, skeletal, or cardiac abnormalities) also have deletions (often submicroscopical) of the terminal portion of the short arm of chromosome 17. Therefore, some researchers suspected that within one deleted region of chromosome 17 was a gene that, when deleted, was sufficient to result in the lissencephaly phenotype. In 1993, the human gene for this form of lissencephaly was identified as LIS1.
The protein encoded by LIS1 has 99% homology to a 45-kD subunit of a bovine brain platelet-activating factor acetylhydrolase (in humans PAFAH1B1). Subsequently, depletion of this protein has been demonstrated in the brains of patients with Miller-Dieker syndrome and with isolated lissencephaly. Interestingly, whereas the message for LIS1 in mice is expressed ubiquitously, the LIS1 protein product has been localized to the neuropile, Cajal-Retzius cells, and
ventricular neuroepithelium at the time of human neuronal migration. However, it is not the lack of enzyme activity that leads to the brain malformation; rather, it appears that the catalytic units of this enzyme complex regulate the available level of LIS1 protein.
ventricular neuroepithelium at the time of human neuronal migration. However, it is not the lack of enzyme activity that leads to the brain malformation; rather, it appears that the catalytic units of this enzyme complex regulate the available level of LIS1 protein.
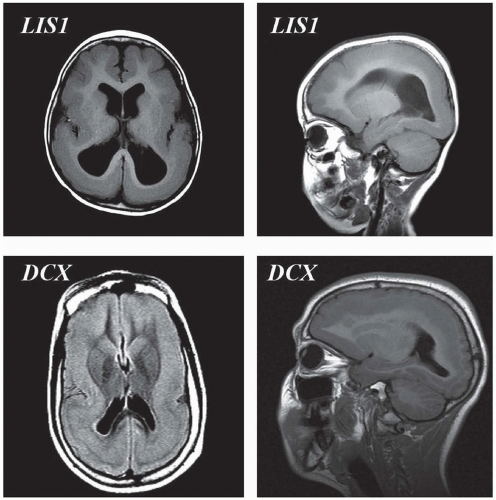 FIGURE 133.5 Lissencephaly spectrum—top panels are from a patient harboring an LIS1 deletion. To the left is an axial T1 magnetic resonance image demonstrating agyria posteriorly and pachygyria anteriorly. To the right is a lateral sagittal T1 magnetic resonance image from the same patient demonstrating the same gradient of posterior worse than anterior malformation. Bottom panels are from a female with a doublecortin (DCX) mutation demonstrating the so-called double cortex (subcortical band heterotopia).
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
