CHAPTER 214 Myelomeningocele and Myelocystocele
Myelomeningocele is a manifestation of a generalized malformation of the central nervous system (CNS) that occurs in 2500 to 6000 newborns per year in the United States.1,2 Its initial management will affect not only the neonates’ survival but also the handicaps that they will have to cope with throughout their lives. Myelocystoceles are uncommon, embryologically unrelated lesions of the distal end of the spinal cord. Although these lesions do not require urgent surgical repair, affected children require lifelong neurosurgical care.
Terminology
Myelomeningocele most likely results from failure of proper neural tube closure during primary neurulation, and such failure leaves a flat plate of neural tissue called the neural placode (Fig. 214-1). As a result, the overlying mesodermal and ectodermal elements fail to form, and an open spinal defect is produced that is almost invariably associated with a Chiari II malformation and its cranial manifestations (Fig. 214-2). A terminal myelocystocele is a skin-covered midline mass composed of a low-lying conus medullaris with a cystic trumpet-like dilation of the caudal central canal, a surrounding meningocele, and a lipoma (Fig. 214-3). These lesions are believed to result from defective cellular differentiation during secondary neurulation. Myelomeningocele and myelocystocele are discussed separately.

Myelomeningocele
Anatomic Considerations
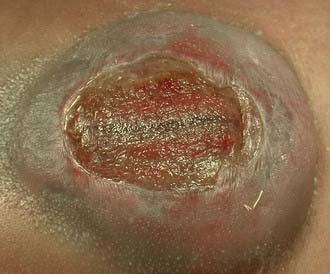
Although failure of closure of the neural tube is thought to be the cause of myelomeningocele, it is likely that other processes, such as errors in gastrulation, may also disrupt neurulation and ultimately produce an open neural tube defect (NTD).3 In all cases of myelomeningocele, failure of neural tube closure results in an exposed neural placode (Fig. 214-4). The groove in the center of the placode is the remnant of the central canal. The spinal roots exit from the anterior surface of the placode such that the ventral roots lie medially and the dorsal roots lie laterally. The dura fuses with the defect in the fascia laterally. Functional neural tissue may be present caudal to the placode or in the nerve roots exiting from the placode.
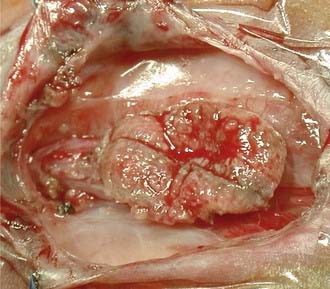
Most myelomeningoceles (85%) are located in the caudal thoracolumbar spine or more distally. Ten percent are in the thorax, and the rest are cervical. Cervical myelomeningoceles are often very similar to meningoceles but without the associated Chiari II malformation and hydrocephalus.4
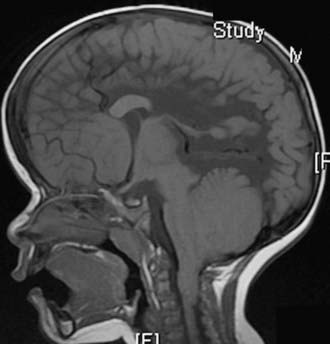
Almost all patients with myelomeningocele also have the Chiari II hindbrain malformation, a constellation of abnormalities that affect the entire CNS (see Fig. 214-2). Associated brainstem defects include medullary kinking, tectal beaking, and intrinsic nuclei abnormalities.5 Supratentorial abnormalities include partial or complete dysgenesis of the corpus callosum, polymicrogyria, a large massa intermedia, and gray matter heterotopia. Mesodermal development of the skull is also affected and leads to a small posterior fossa, short clivus, low-lying tentorium and torcular Herophili, wide incisura, and enlarged foramen magnum. Lückenschädel, or craniolacunia (scalloping of the skull bones noted on computed tomography [CT] and plain radiographs of the skull in these infants), is also a result of this mesodermal dysplastic process; these skull lacunae are not caused by increased intracranial pressure and usually resolve by 1 year of age.
The majority (80% to 90%) of patients with myelomeningocele have hydrocephalus that requires treatment. The hydrocephalus may result from both obstructive and communicating components. Syringomyelia occurs in 40% to 80% of patients with spina bifida and is usually nonprogressive.6 In patients with spina bifida, neurological deterioration can result from symptomatic hydrocephalus, Chiari II malformation, syringomyelia, or retethering. Most often, the cause of neurological deterioration is hydrocephalus from malfunction of a shunt.6
History
There is evidence that spina bifida existed in ancient civilizations.7 Peter Van Forest first recorded a child with spina bifida in 1587,8 and in 1610 he performed the first reported surgical resection of the myelomeningocele sac.9 The first anatomic illustration was drawn by Tulp in 1641.10 In 1761, Morgagni was the first to associate the clinical changes observed in spina bifida patients with the myelomeningocele.11 The first theory to explain spina bifida was advanced by Lebedeff in 1881.12 He attributed myelomeningocele to failure of the neural tube to close. In 1886, von Recklinghausen described the types of spina bifida and reviewed the surgical treatment.13 In Fraser’s report of the first series of spina bifida patients treated surgically, two thirds of the patients operated on between 1898 and 1923 survived until hospital discharge. Six years later, nearly a quarter (23%) of the patients were still alive.14
More aggressive surgical treatment for children with spina bifida was undertaken after the development of ventriculoperitoneal shunting for hydrocephalus in the 1950s. Because delayed complications developed in many patients, some physicians suggested selective surgical treatment of neonates with spina bifida.15,16 The ethical debate about selective treatment ended in the early 1980s when reports demonstrated that patients in nonselected series did as well as or better than those in series with selection.17 In North America, unless there is an anomaly incompatible with life, almost all newborns with myelomeningocele are treated aggressively to optimize their quality of life.
Epidemiology
In the United States, the prevalence of myelomeningocele has declined because of both prenatal folate supplementation and termination of pregnancy. Before 1980, the prevalence of myelomeningocele in the United States was 1 to 2 per 1000 live births. More recently, the prevalence has declined to 0.44 per 1000 live births.18 Twenty percent to 30% of the decline can be attributed to pregnancy termination after prenatal diagnosis.19–21
Geographic variation in the prevalence of spina bifida has been recognized for many years. The United Kingdom, particularly Ireland, has a higher prevalence of NTDs than do continental Europe and the United States. The prevalence of spina bifida in the United Kingdom after 1980 was 0.74 to 2.5 per 1000 live births; the prevalence in the United States during the same period was 0.41 to 1.43 per 1000 live births.22
Racial and ethnic variation in the prevalence of spina bifida exists and persists after immigration. In the United States, the prevalence of NTDs is highest in Hispanics, followed by whites and then African Americans and Asians. The overall U.S. prevalence was 0.46 per 1000 live births, but Hispanics had a prevalence rate of 0.6 per 1000 and Asians 0.23 per 1000.18 Internationally, the prevalence of spina bifida varies from a low of 0.1 per 1000 live births in native Africans to a high of 12.5 per 1000 in Celtics.22 Patterns of variation in the severity of the defect also exist among ethnic and racial groups. With regard to gender differences, a slight (0.57% to 0.71%) female preponderance exists.23
Pathogenesis
Closure of the posterior neuropore occurs during human embryonic stage 12, at approximately 26 days of gestation. Although the etiology of some types of open NTDs may be related to errors in gastrulation,3 the most widely supported theory of spina bifida formation proposes that the posterior neuropore fails to close completely during neurulation and a myelomeningocele, Chiari II malformation, and hydrocephalus result. The nonclosure theory has been substantiated by recent experimental studies using toxic agents and animal mutants. Toxic agents include cytochalasin, vinblastine, calcium-channel antagonists, phospholipase C, concanavalin A, retinoic acid, hydroxyurea, and mitomycin C. Folate and its pathophysiology have been the focus of the majority of recent investigations. Mutant mice, such as the splotch and curly tail/loop tail mouse,24 have been used to study the pathogenesis of NTDs.
A unified hypothesis to explain the sequential development of myelomeningocele and Chiari II hindbrain malformation has been proposed.25 In normal development, during the period of rapid brain enlargement that occurs after closure of the posterior neuropore, the neurocele (primitive central canal) becomes transiently occluded. If the posterior neuropore has failed to close, thereby causing a myelomeningocele, the neurocele fails to occlude. Without appropriate neurocele occlusion, cerebrospinal fluid (CSF) flows out through the defect in the open posterior neuropore. With the lack of distention of the brain by CSF, formation of the cranium and its contents is disrupted. The posterior fossa is small, and both upward and downward herniation of the cerebellum occurs and results in the Chiari II malformation.25 The unified hypothesis has been supported by experiments with chick embryos and with the homozygote splotch mouse.
Etiology
Folate
The coenzyme folate is required for hematopoiesis, metabolism, and normal gastrointestinal and neurological function. The association between folate insufficiency and NTDs was suspected by Hibbard in 1964. A randomized, double-blind study in 1991 demonstrated that couples in the United States with a previous history of an infant with an NTD have a 2% to 3% chance of having a second child born with an NTD. If 4 mg of folic acid was consumed during the critical period (before and during pregnancy), the risk of having a second child born with spina bifida dropped 71%,26 thus suggesting that some, but not all NTDs are related to abnormal folate supplementation (Table 214-1).27 Current recommendations state that women who could potentially become pregnant should consume at least 0.4 mg/day of folic acid (Table 214-2).28 The cause of the NTDs that are not prevented by folate supplementation is not known.
TABLE 214-1 Risk Factors for Neural Tube Defects
| RISK FACTORS | RISK (%) |
|---|---|
| Medical | |
| History of pregnancy with an NTD | 2-3 |
| Partner with an NTD | 2-3 |
| Diabetes mellitus type 1 | 1 |
| Seizure disorder (valproic acid or carbamazepine) | 1 |
| Close relative with an NTD | 0.3-1 |
| Prepregnancy obesity (>110 kg) | 0.2 |
| Nonmedical | |
| Agricultural pesticides and chemicals | |
| Cleaning solvents and disinfectants | |
| Nursing | |
| Radiation exposure | |
| Anesthetic agents | |
| Hot tubs, saunas, and fever (hyperthermia) | |
| Lead | |
| Tobacco smoke | |
NTD, neural tube defect.
Adapted from Cohen AR, Robinson S. Myelomeningocele: Early management. In: McLone DG, ed. Pediatric Neurosurgery, 4th ed. Philadelphia: WB Saunders; 2000:241-259.
TABLE 214-2 Recommendations for Daily Folate Supplementation
| CIRCUMSTANCE | DOSE (mg) |
|---|---|
| Before Pregnancy | |
| Women with no known risk factor | 0.4 |
| Women at high risk | 4 |
| During Pregnancy | |
| Women with no known risk factor | 0.6 |
| Women at high risk | 4 |
| Post Partum While Breast-Feeding | |
| Women with no known risk factor | 0.5 |
| Women at high risk | 4 |
Adapted from Cohen AR, Robinson S. Myelomeningocele: Early management. In: McLone DG, ed. Pediatric Neurosurgery, 4th ed. Philadelphia: WB Saunders; 2000:241-259.
Anticonvulsants
NTDs are associated with antiepileptic medications taken during pregnancy. The risk of a mother treated with carbamazepine or valproic acid having a child with spina bifida or anencephaly is 1% or 1% to 2%, respectively.29
Diabetes Mellitus
Type 1 diabetes mellitus places woman at higher risk (1%) for a pregnancy complicated by spina bifida. This elevated risk appears to be independent of the increased risk for an NTD from prepregnancy obesity.2
Prenatal Diagnosis
Diagnostic Studies
Maternal Serum Alpha Fetoprotein
Determination of maternal serum alpha fetoprotein (MSAFP) levels in the early part of the second trimester is the initial screen for NTDs. In amniotic fluid, the AFP concentration is 100-fold less than in fetal CSF.32 The optimal time for sampling MSAFP is at 16 to 18 weeks, although MSAFP levels can be determined between 14 and 21 weeks’ gestation. Seventy-nine percent of pregnancies with open NTDs and 3% of normal singleton pregnancies have an MSAFP level that is 2.5 multiples of the median (MoM) at 16 to 18 weeks’ gestation.33 Based on the adjusted MoM MSAFP level for the patient’s age and population, the MSAFP level at a specific gestational age provides a patient-specific risk for an NTD. For example, an MSAFP level of 1.5 MoM predicts a risk of 1 in 2317 for an open NTD, and an MSAFP level of 2.5 MoM predicts a risk of 1 in 98. The estimated risk is adjusted according to the prevalence of NTDs in the population. The diagnostic accuracy of a single MSAFP level is limited to 60% to 70%. If the risk for spina bifida predicted by the MSAFP concentration is greater than 1 in 500, further testing with high-resolution ultrasonography is recommended.
If the screening MSAFP level is elevated, the test can be repeated. A constant or decreasing MSAFP level is not consistent with a pregnancy with an NTD. In 40% of cases, the repeated level is normal.34 Instead of repeating the test, high-resolution fetal ultrasonography can be performed. Ultrasonography can identify false-positive MSAFP levels caused by incorrect estimated gestational age, multiple pregnancy, or fetal demise. If the initial and repeated MSAFP levels are elevated more than 3 MoM and the first fetal ultrasound study is normal, a second ultrasound study is recommended.
Ultrasonography
The sensitivity of high-resolution fetal ultrasonography is nearly 100% in prenatal screening for NTDs. An accurate prediction of the anatomic level of the spinal cord defect can be obtained in 64% of cases and is within one level of the defect in 79%.35 High-resolution ultrasonography can also visualize two cranial abnormalities that are characteristic of hydrocephalus and the Chiari II malformation associated with spina bifida. The first, consisting of scalloping of the frontal bones on a biparietal view, is called the lemon sign.36,37 The second, consisting of an abnormally shaped midbrain, elongated cerebellum, and obliteration of the cisterna magna characteristic of the Chiari II malformation, is referred to as the banana sign. The lemon sign was present in 80% of fetuses with myelomeningocele, and the banana sign was present in 93%.36,37
The correlation among the findings of prenatal ultrasonography, level of spina bifida, degree of posterior fossa deformity, and degree of hydrocephalus has been examined.38 No correlation was found between the ventricular diameter and the level of the spinal defect.39,40 The posterior fossa deformity did not progress on serial ultrasound examinations.38 If ultrasonography is nondiagnostic, magnetic resonance imaging (MRI) or amniocentesis may be indicated.
Magnetic Resonance Imaging
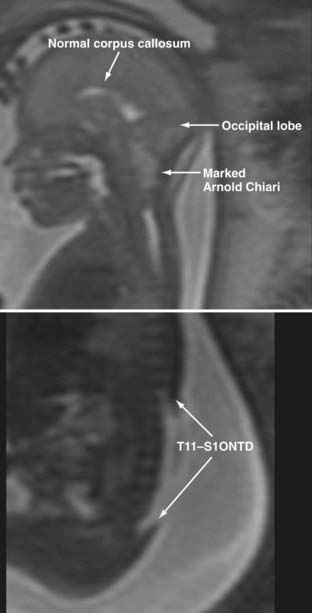
Prenatal MRI is increasingly being used to examine potential fetal neurological anomalies (Fig. 214-5), and it provides greater resolution than ultrasonography.41 Because the recent development of fast spin echo techniques has largely overcome the challenges associated with fetal movement, MRI has become an excellent noninvasive second-line imaging modality for cases in which screening high-resolution ultrasonography is nondiagnostic. Data regarding rates of diagnostic accuracy in detecting myelomeningocele are not currently available.
Amniocentesis
Amniocentesis may be indicated if the MSAFP level and imaging studies suggest the presence of an NTD. The amniotic acetylcholinesterase (AChE) level is used to increase the diagnostic accuracy of the amniotic AFP level because the latter can have a high false-positive rate in a population at low risk for NTDs.42 If an NTD is present, neural AChE from CSF leaks into the amniotic fluid. Other anomalies such as an omphalocele or Turner’s syndrome can elevate the amniotic AChE level.43 Amniotic fluid AFP and AChE levels had an accuracy of 99% and a false-positive rate of 0.34% in a large study of singleton pregnancies.44
Differential Diagnosis
At least 22 other fetal abnormalities besides myelomeningocele increase MSAFP levels.32 Abdominal abnormalities such as omphalocele, cloacal exstrophy, esophageal atresia, annular pancreas, duodenal atresia, and gastroschisis and urologic abnormalities such as congenital nephrosis, polycystic kidneys, urinary tract obstruction, and renal agenesis can also elevate MSAFP levels.33,42 Elevated MSAFP and elevated amniotic fluid AFP and AChE can be caused by a sacrococcygeal cystic teratoma, but the teratoma can usually be distinguished from a myelomeningocele by fetal ultrasonography.
Prenatal Counseling
Prognosis
The present 2-year survival rate of neonates born with myelomeningocele is greater than 95%. Ten percent to 15% of children with spina bifida die before 6 years of age, even with aggressive treatment.1,45 Hydrocephalus that requires treatment occurs in most (90% to 98%) of the patients.1,45
The functional motor level determines the ambulatory status of a patient. The motor level may or may not correspond to the anatomic level of the myelomeningocele. The prenatal anatomic level determined by ultrasonography accurately predicts the motor function level at the age of 3 to 4 years better than does determination of the neonatal motor level by examination.46 L3 function allows one to stand erect, and L4 and L5 function allows ambulation.47 During the first decade, approximately 60% of children with spina bifida are community ambulators, without or with assistive devices (including wheelchairs); 15% are household ambulators and 26% are nonambulatory.48 The percentage of community ambulators decreases by about 17% during adolescence. As teenagers, many patients with spina bifida have to increase the level of assistance provided by devices. Normal urinary continence is present in only a small portion (6% to 17%) of these children.47 Most (85%) are socially continent by using a combination of clean intermittent catheterization and medications, and most (86%) have social fecal continence.49
The majority (75% to 80%) of children with spina bifida can have normal intelligence with aggressive management of hydrocephalus and infections. A correlation between a higher myelomeningocele level or severe ventricular enlargement on prenatal ultrasonography and lower intelligence has been found in some studies.17,50,51 The intelligence quotient (IQ) remained normal if the CNS was not infected, but only about a third (31%) of the patients with a history of CNS infection had a normal IQ.52 It remains controversial whether the number of shunt revisions affects IQ.50,53 Although complex learning disabilities are not unusual in children with spina bifida, more than half (58%) of children with spina bifida perform at their grade level.49 Because of severe cognitive impairment, 10% to 15% require custodial care.1 Only a small portion (<10%) of patients with spina bifida become economically independent. The majority (>80%) of patients with spina bifida need psychiatric care or counseling to help them cope with their disabilities.
Risk in Siblings
The parents of a child with a myelomeningocele may seek counseling about the chance of having another child with an NTD. The risk of a sibling having an NTD is estimated to be 2.8% in the United States.54 If the parents have had two previous pregnancies with NTDs, the risk increases to 4.8%.55 If a parent has an NTD, the risk of a child having spina bifida is 3%.56 Monozygotic twins have a low concordance rate.57 Autosomal dominant, autosomal recessive, and X-linked inheritance patterns have been described.
Fetal Management of Spina Bifida and Hydrocephalus
A role for intrauterine surgery to minimize the neurological deficits of patients with myelomeningoceles has been suggested but remains highly controversial. According to some proponents of intrauterine myelomeningocele repair (IUMR), a portion of the neurological deficit present in children with myelomeningoceles is acquired from persistent exposure of the dysplastic spinal cord to amniotic fluid.58 In experimental animal models, intrauterine repair to cover the exposed spinal cord restored normal development.59–62 Other experiments in rodents have shown that the dysplasia of the dorsal spinal cord results from the lack of normal afferent input from aberrant dorsal roots rather than from deterioration of normal tissue as a result of exposure to amniotic fluid.63
Recent studies have shown that IUMR does not improve the motor deficit, but it may minimize the degree of cerebellar herniation in the Chiari II malformation. In a nonrandomized observational study, patients who underwent IUMR were compared with patients who underwent standard neonatal repair.64 The infants undergoing intrauterine repair were less likely than the control infants to require a shunt for symptomatic hydrocephalus during the first 6 months (59% versus 90%), and they demonstrated less hindbrain herniation (38% versus 95%). It is, however, important to note that this was an uncontrolled, observational study. The infants who underwent intrauterine repair were born significantly earlier than were the control infants (33 versus 37 weeks’ gestation). Further studies have suggested that greater rates of prematurity translate into an increased risk for fetal morbidity and mortality.65 The long-term effects on the cognitive development of children who have undergone IUMR have yet to be assessed, although studies of short-term neurodevelopmental outcomes indicate that those who do not require shunting may have improved outcomes.66 Data with regard to urologic outcomes after IUMR are also inconclusive at this time.67 To determine whether IUMR is beneficial when compared with conventional postnatal closure of myelomeningocele, the Management of Myelomeningocele Study (MOMS) was initiated in 2003. MOMS is a prospective, randomized trial that is being completed at three U.S. centers. The primary end points include death and shunt placement. Secondary outcomes include leg function, bowel and bladder function, and intelligence, among others. MOMS was scheduled for completion in 2009.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


