Chapter 56 Myoclonic Seizures and Infantile Spasms
Introduction
Myoclonus, myoclonic seizures, and infantile spasms share many common features yet are seen in a wide variety of neurologic conditions. Myoclonus is not a diagnosis, but rather a sign that can have many underlying etiologies. The precise definitions of each of these terms has also been somewhat controversial, but for the purpose of this chapter we will use the definition proposed by Victor and Adams [Ropper and Samuels, 2009] and define myoclonus simply as a “shocklike irregular jerk,” with myoclonic seizures having a similar clinical appearance but also accompanied by neurophysiologic evidence of being cortically generated. While these definitions each have their own potential flaws, the distinction between a cortically generated movement (myoclonic seizure) and a subcortically generated movement (myoclonus) is extremely important, especially when thinking about some of the progressive myoclonic epilepsies, as many of these patients can manifest both myoclonus and myoclonic seizures. Infantile spasms, however, should not be confused with either, as the spasm itself is often a more complex constellation of movements with a myoclonic component.
Epilepsy Syndromes with Prominent Myoclonic Seizures
Myoclonic seizures can occur in a wide variety of pediatric epilepsy syndromes, including Lennox–Gastaut syndrome and childhood absence epilepsy, which are discussed elsewhere in Part VIII. Our focus will be on those syndromes in which myoclonus is a critical feature for the diagnosis. This coverage includes benign myoclonic epilepsy in infants (BME), severe myoclonic epilepsy in infancy (SMEI/Dravet’s syndrome), idiopathic epilepsy with myoclonic-astatic seizures (IEMAS), and juvenile myoclonic epilepsy (JME) (Table 56-1).
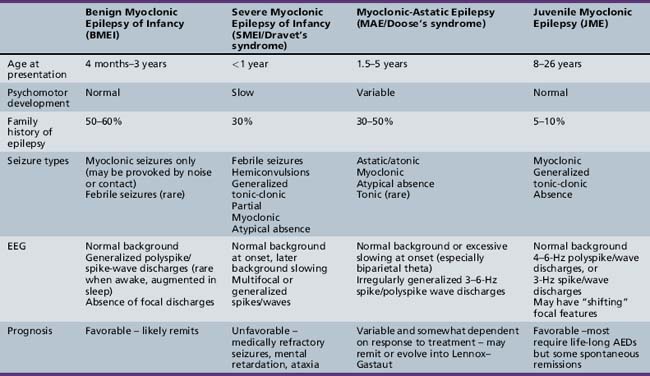
Benign Myoclonic Epilepsy of Infancy
BMEI was first described by Dravet and Bureau in 1981 [Dravet and Bureau, 1981]. The syndrome is characterized by brief, generalized myoclonic seizures, with the predominant area of muscle involvement being the proximal upper extremities [Hirano et al., 2009], and usually occurs in children between the ages of 4 months and 3 years, although cases have been described with onset up to 4 years 8 months [Rossi et al., 1997]. These attacks often occur multiple times per day. Detailed electroencephalography (EEG) and polygraphic electromyography (EMG) recordings have shown that the myoclonic seizures are often associated with a flexor postural change and approximately 80 percent of attacks involve the upper limbs [Hirano et al., 2009]. A history of febrile seizures (30 percent) and a family history of epilepsy (39 percent) is relatively common [Roger et al., 2002].
EEG Findings
The ictal EEG discharge is often a generalized spike wave (GSW) that may be slower than 3 Hz [Hirano et al., 2009]. Occasionally, the myoclonic seizures can be massive and result in a fall, or they may occur in a cluster. Some infants may exhibit photosensitivity at the onset of the syndrome, and the photic-induced myoclonic seizures are often more prominent than those that occur spontaneously [Capovilla et al., 2007]. Other infants may be sensitive to acoustic or tactile stimuli, leading some to argue for a distinctly separate epilepsy syndrome [Ricci et al., 1995]. We and others [Roger et al., 2002] do not believe this is a clinically distinct syndrome, but rather that the syndrome of BME may include both photosensitive and stimulus-provoked seizures in addition to spontaneous myoclonic seizures. In both groups, the waking EEG in between seizures is often normal but may contain rare generalized spike/wave discharges [Ricci et al., 1995; Capovilla et al., 2007], and the ictal EEG findings are indistinguishable.
Treatment and Outcome
The overall prognosis of myoclonic seizures is excellent, with complete resolution in almost all children usually within 1–2 years of diagnosis. In those who demonstrate photosensitivity, these seizures may be more difficult to control and may persist for a longer period of time [Roger et al., 2002]. The majority of children also have normal neurodevelopmental outcomes [Caraballo et al., 2009], although some studies have reported impaired psychomotor development and behavioral disturbances if the child is not treated or if onset of the syndrome is at less than 2 years of age [Mangano et al., 2005]. Those children with a prominent reflex component may have a more favorable neurodevelopmental outcome [Zuberi and O’Regan, 2006].
The medication of choice appears to be valproic acid (VPA), with 80–90 percent responding to VPA monotherapy, although high serum levels (>100 mg/L) may be necessary. When VPA monotherapy does not provide complete control, adjunctive use of a benzodiazepine, such as clonazepam or clobazam, can be helpful [Rossi et al., 1997].
Severe Myoclonic Epilepsy in Infancy
First described in 1978 by Charlotte Dravet, SMEI occurs in normally developing children who experience prolonged febrile (>20 minutes) or afebrile seizures, including hemiconvulsions, during the first year of life. Afebrile, mixed seizures follow, and the emergence of myoclonic seizures is common but not necessary for the diagnosis (borderline variant). For this reason, the syndrome is better referred to as Dravet’s syndrome, as the name severe myoclonic epilepsy of infancy may mislead clinicians and cause them not to suspect the diagnosis if myoclonic seizures are absent. The syndrome was once thought to be exceedingly rare, with an incidence of 1 in 40,000 children [Hurst, 1990], but with heightened awareness of the clinical spectrum and the availability of genetic testing, this may be an underestimation of the true incidence.
Mutations in the SCN1A Gene
The importance of SCN1A gene mutations as an underlying etiology in epilepsy first became apparent when Scheffer and Berkovic reported missense mutations in two families with the syndrome of generalized epilepsy with febrile seizures plus (GEFS+) [Scheffer and Berkovic, 1997]. Due to the common occurrence of febrile seizures in both GEFS+ and Dravet’s syndrome, Claes et al. screened seven patients with Dravet’s syndrome and found de novo missense mutations in all seven patients [Claes et al., 2001]. Testing for mutations in the SCN1A gene is clinically available, and hundreds of patients with Dravet’s syndrome with SCN1A mutations have been reported [Depienne et al., 2009]. The main difference between individuals with a GEFS+ phenotype and a Dravet phenotype appears to be that the former often have a missense mutation with reduced penetrance, whereas patients with Dravet have mutations that occur in isolated patients (arise de novo) [Claes et al., 2001].
Mutations in the SCN1A gene are seen in approximately 70–85 percent of patients with Dravet’s syndrome [Harkin et al., 2007], and therefore it remains a clinical diagnosis based on age of onset and clinical phenotype. One should not exclude the diagnosis on the basis of a negative result on SCN1A mutation testing. Approximately 200 different point mutations in the SCN1A gene were identified in 271 probands with a strict clinical diagnosis of Dravet’s syndrome [Depienne et al., 2009]. Of those probands without a point mutation, the multiplex ligation-dependent probe amplification (MPLA) technique found an additional 14 micro-rearrangements of SCN1A [Depienne et al., 2009]. An SCN1A variant database was published in 2009, listing each type of mutation and the associated epilepsy syndrome [Claes et al., 2009]. As of 2009, 648 point mutations have been reported, of which 582 (90 percent) are in subjects with Dravet’s syndrome. An additional 67 genomic rearrangements were reported, with 46 (7 percent) of these occurring in patients with Dravet’s syndrome. The remaining clinical phenotypes with SCN1A mutations are wide and varied [Harkin et al., 2007]. In Dravet’s syndrome, the end result of each of these mutations appears to be a total loss of function of the mutated allele, most commonly caused by a missense mutation that leads to an abnormality in the pore-forming unit of the sodium channel [Claes et al., 2009]. While specific mutation genotype–phenotype correlations within Dravet’s syndrome are not yet available, further research in this area may lead to particular treatment strategies for specific genotypes.
Seizure Semiologies
Patients with Dravet’s syndrome have many different seizure semiologies, often occurring in the same patient. The most common presenting seizure type is a prolonged febrile convulsion lasting longer than 25 minutes, but afebrile seizures can be the presenting seizure type in as many as 35 percent [Roger et al., 2002]. Japanese authors have observed that hot water immersion can trigger seizures in these patients [Fujiwara et al., 1990], and this feature is one part of a clinical screening test aimed at predicting the diagnosis of Dravet’s syndrome [Hattori et al., 2008]. Other convulsive seizure types include generalized tonic-clonic seizures, hemiclonic seizures, and “falsely generalized,” seizures as described by Dravet [Roger et al., 2002]. The latter has a complex semiology, with a high degree of discrepancy between the clinical and EEG findings. Seizures often start in one part of the body, spread to another (on the opposite side), and then potentially return to the original side of origin, only to involve a new body part.
Nonconvulsive seizures are also common and include simple partial, complex partial, atypical absence, and myoclonic types. The specific semiologies of each of these seizure types are similar to that seen in other forms of childhood epilepsy. Tonic seizures are rare. A unique seizure type, termed “obtundation status,” is unique to the syndrome. The semiology of this seizure type consists of variable impairment in consciousness, and fragmentary and erratic segmental myoclonias involving the limbs and face. Patients may still be able to engage in simple activities, such as eating or playing with a toy. These episodes may last several hours to days and can either be initiated by, or conclude with, a convulsion [Roger et al., 2002]. The EEG during these episodes is not a pattern of classical atypical absence status, but rather is characterized by a diffuse dysrhythmia with focal or diffuse spikes [Roger et al., 2002]. We have observed one patient, who would begin with this type of clinical phenomenon, including prominent eyelid myoclonia that was dramatically accentuated with eye closure, and would remain in this state for 2–3 days, culminating in a generalized convulsion, after which her mental status cleared. She would remain clear for 2–3 weeks, and then repeat the same cycle over again.
EEG Findings
The EEG is invariably normal early in the course of the disease, and therefore, in contrast with other epilepsy syndromes, the diagnosis must be suspected on clinical grounds. As the disease progresses, the EEG background becomes slow but often continues to manifest normal sleep architecture [Roger et al., 2002]. Some authors have observed rhythmic 5–6-Hz centroparietal theta but this is not specific to the syndrome [Doose et al., 1998]. Over time, the interictal EEG can show both generalized and multifocal spikes, or may not show any of these features despite frequent seizures of multiple types, further highlighting the lack of specificity of EEG findings. Given this lack of specificity, a screening test for predicting the diagnosis of Dravet’s syndrome under 1 year of age has been developed, with a positive predictive value as high as 94 percent [Hattori et al., 2008]. This screening test uses simple clinical features, as shown in Table 56-2. A sum score of 6 or greater is associated with a high risk of having Dravet’s syndrome, and therefore SCN1A testing should be done in those cases. Even if the hot water-induced risk factor is excluded, a score of 5 or higher is still indicative of a high-risk patient.
Table 56-2 Clinical Screen for Dravet’s Syndrome
| Clinical Score | Risk Score |
|---|---|
| Onset <7 months | 2 |
| More than 5 seizures prior to 12 months of age | 3 |
| Hemiconvulsion | 3 |
| Focal seizure | 1 |
| Myoclonic seizure | 1 |
| Prolonged seizure (>10 minutes) | 3 |
| Hot water-induced seizure | 2 |
(Adapted from Hattori J et al. A screening test for the prediction of Dravet syndrome before one year of age. Epilepsia 2008;49(4):626–633.)
Imaging
Magnetic resonance imaging (MRI) studies are usually normal but may show nonspecific cerebral or cerebellar atrophy or isolated ventricular enlargement. Interestingly, these findings, specifically diffuse brain atrophy, appear to be more common in those children without associated SCN1A mutations. Furthermore, despite having frequent prolonged febrile and afebrile convulsions, mesial temporal sclerosis is rare, reported in only 1 of 58 patients [Striano et al., 2007].
Treatment
Until recently, treatment strategies were generally unsuccessful, with poor seizure control despite polytherapy. Many have observed that certain antiepileptic drugs (AEDs) exacerbate seizures, with the most notorious agents being carbamazepine (CBZ) and lamotrigine (LTG) [Wakai et al., 1996; Guerrini et al., 1998]. Some authors have recommended using CBZ as a means of “confirming” the diagnosis when there is a high index of clinical suspicion [Wakai et al., 1996]. Given the availability of genetic testing, we do not endorse this practice.
A study by Chiron et al. and a subsequent meta-analysis pooling these data with unpublished data showed that combination therapy with valproate (VPA) + stiripentol (STP) + clobazam (CLB) resulted in a 70 percent decrease in seizure frequency, with 43 percent of patients becoming seizure-free [Chiron et al., 2000; Kassai et al., 2008]. In this study, the STP dose began at 50 mg/kg/day divided in 2–3 doses, but could be titrated up to 100 mg/kg/day. The maximum recommended dose is 3500 mg/day [Chiron et al., 2000].
One proposed mechanism for the effect of STP is the potent inhibition of the p450 cytochrome system, resulting in higher levels of VPA and CLB. Plasma levels of CLB were significantly higher but levels of VPA were not [Chiron et al., 2000]. Interestingly, one other result of p450 inhibition is lower levels of metabolites, some of which are thought to explain some of the adverse toxic effects of AEDs. This finding perhaps explains why patients are able to tolerate the higher doses of certain AEDs [Chiron, 2005]. These observations, however, raise the possibility that STP has other mechanisms of action independent of its effects on the p450 system, including enhancement of gamma-aminobutyric acid (GABA)ergic transmission [Quilichini et al., 2006].
Of the newer agents, topiramate (TPM) appears to be very effective, especially if added to STP, resulting in 50 percent reduction of seizures in 78 percent of patients at a modest optimal dose of 3.2 mg/kg/day. Seventeen percent of patients remained seizure-free for at least 4 months [Kroll-Seger et al., 2006].
Bromide therapy has also shown promise in several studies [Oguni et al., 1994], specifically in reducing the number of convulsions. The ketogenic diet may be helpful in some patients [Caraballo et al., 1998].
Aggressive treatment of acute seizures and prevention of status epilepticus is critical, as some small studies have shown a trend towards improved neuropsychologic outcomes in those children with less frequent convulsive seizures [Chipaux et al., 2008]. Each patient should have a clearly defined “emergency plan,” and the clinician should ask each family if a particular treatment regimen has been particularly effective for their child [Nolan et al., 2006]. Parents should be educated about the inevitability of seizures, especially prolonged seizures in the setting of fever. Early use of high doses of benzodiazepines, either in the form of rectal diazepam (as high as 1 mg/kg per dose) or buccal midazolam, should be the first-line therapy. Some physicians have recommended insertion of a central venous port for those children who have proven to have difficult intravenous access and multiple episodes of status epilepticus [Dooley et al., 1995]. In these specific cases, families are able to cope much better, knowing that multiple intravenous attempts will not be necessary and appropriate treatment can be initiated to terminate the seizure faster.
Outcome
Long-term outcome with regard to seizure control and neuropsychological development is variable but generally poor. By definition, all children with Dravet’s syndrome have normal development at onset. Around 50 percent of children walk unsupported by a mean of 16 months of age; however, it is rare for children to utilize two-word sentences at the normal age of around 2 years [Wolff et al., 2006]. At around 2 years of age, there appears to be a gradual decline, and then there is a relative stabilization between the ages of 4 and 6 years [Wolff et al., 2006]. A trend was noted that fewer convulsive seizures often resulted in higher developmental quotients. Subsequent small cohorts have reported patients with normal or near-normal IQs and this favorable outcome is attributed to early diagnosis and appropriate management [Chipaux et al., 2008]. It remains to be seen in larger prospective studies whether early diagnosis and appropriate management with the medications outlined above will lead to improved seizure control and a subsequent improvement in neurodevelopmental outcome.
Myoclonic-Astatic Epilepsy of Doose
Etiology
Genetic factors clearly play a role in the pathogenesis of MAE, based on the high incidence of seizures (32 percent) in probands’ siblings and parents; the most common seizure type is absence seizures. Despite the initial discovery of the SCN1A gene in a family with GEFS+ that included a family member with MAE [Scheffer and Berkovic, 1997], only a small number of patients with mutations in the SCN1A gene have been identified in patients with MAE [Ebach et al., 2005; Harkin et al., 2007]. Further genotype–phenotype correlation studies are necessary to determine if there are underlying genetic factors that would be able to predict response to treatment and outcome.
Seizure Semiologies
Onset of MAE is often between the ages of 2 and 6 years, and children are usually developmentally normal [Kaminska et al., 1999]. These children may begin experiencing unexplained “jerks” and “falls,” which occur multiple times per day. These myoclonic seizures can take place in isolation, but often are followed by a brief period of atonia that results in a dramatic fall in which the child appears to be propelled to the ground. Polygraphic studies have documented that atonic falls can occur with or without the preceding myoclonus, and occur in about 64 percent of patients [Oguni et al., 2002]. Although atonic falls are not necessarily seen in all patients, more subtle myoclonic/atonic seizures are common. The only manifestation of these may be a brief head nod or “head drop,” with or without a myoclonic jerk of the upper extremities. When head drops occur in clusters with an associated alteration in level of consciousness, nonconvulsive status should be suspected. EEG findings during these episodes are always abnormal, with frequent runs of generalized spike/wave and slow spike-and-wave complexes. Absence seizures are quite common and may be similar to those seen in typical childhood absence epilepsy, but often are accompanied by more prominent myoclonus of the proximal upper extremities.
Treatment
Early recognition of the syndrome allows treatment to be focused on the broad-spectrum agents that have been shown to be particularly effective. The most commonly used and most effective AEDs have been VPA, LTG, levatiracetam, TPM and various benzodiazepines (clonazepam, clobazam, clorazepate). When absence seizures are frequent, ethosuximide (ETX) can be effective and has also been reported to decrease the frequency of myoclonic seizures; however, it is rarely used as monotherapy for this indication [Oguni et al., 2002]. The combination of VPA/ETX may be particularly effective, especially if high doses of VPA are used.
In those children who are refractory to medical management, a trial of the ketogenic diet should be considered early. The ketogenic diet has been shown to be as effective as other first-line agents in children with MAE [Kilaru and Bergqvist, 2007], and given this efficacy, we consider the ketogenic diet early in our treatment algorithm, especially if there is associated neurodevelopmental decline.
Outcome
The outcome with regard to cognitive development is highly variable but appears to be somewhat dependent on degree of seizure control. Some children may have fairly frequent seizures of multiple types and still have spontaneous remission, usually after approximately 3 years [Kaminska et al., 1999; Oguni et al., 2002]. In some children, the multiple seizure types remain intractable and these children have a higher risk of both cognitive and behavioral disturbances [Oguni et al., 2002]. The occurrence of tonic seizures has been described as a negative prognostic sign, although it remains unclear if these children may actually have an atypical form of Lennox–Gastaut syndrome rather than an atypical form of MAE [Kaminska et al., 1999].
Juvenile Myoclonic Epilepsy
JME is an idiopathic generalized epilepsy (IGE) syndrome characterized by myoclonic seizures, generalized tonic-clonic seizures, and absence seizures. It is extremely common, accounting for 26 percent of IGE and 10 percent of all epilepsies [Janz and Durner, 1998], although this may still be an underestimation; it is very likely underdiagnosed, as many clinicians may not inquire about the presence of myoclonic seizures [Renganathan and Delanty, 2003]. Mean age at onset is highly variable and difficult to determine truly, as some children with childhood absence epilepsy may evolve into the syndrome of JME. Excluding this population, the average age of onset is 15.1 years (7–28 years), with a slight female predominance [Martinez-Juarez et al., 2006].
Seizure Semiologies
In the classic syndrome of JME, myoclonic seizures may precede the first generalized tonic-clonic (GTC) seizure by 6–12 months, although GTCs occur as the first seizure type in approximately one-third of patients [Martinez-Juarez et al., 2006]. Some have proposed specific subgroups of JME, separating those patients who present with typical childhood absence or juvenile absence seizures, although this is much less common than the classical presentation of JME, accounting for only 10 percent of cases. The impact this distinction has on outcome will be discussed below. Photosensitivity is relatively common, occurring in approximately 30 percent of patients [Zifkin et al., 2005].
Myoclonic seizures often occur in the early morning hours shortly after awaking. Transcranial magnetic stimulation studies in patients with JME have demonstrated increased cortical excitability that is not present in other patients with focal epilepsy [Badawy et al., 2006]. Provocation of myoclonic seizures has been linked to higher cognitive tasks requiring higher-order thinking, such as writing or written calculation [Matsuoka et al., 2000].
Specific lifestyle features have been commonly associated with breakthrough seizures, including sleep deprivation, stress, alcohol, and menses [Martinez-Juarez et al., 2006].
EEG Findings
Patients with JME often have abnormal interictal EEGs, with the most common finding being generalized 4–6-Hz polyspike-and-wave (61 percent) or 3-Hz spike-and-wave discharges (14 percent) [Martinez-Juarez et al., 2006]. More prolonged (1–6 days; average 1–2 days) video EEG studies have shown EEG abnormalities of JME in 88 percent of patients [Park et al., 2009]. Other series have reported on the increased yield of an early morning EEG compared to an afternoon EEG (generalized epileptiform activity in 69 percent and 25 percent, respectively) [Labate et al., 2007]. A photoparoxysmal response in treatment-naive patients is also common and has been seen in as many as 35 percent of patients [Specchio et al., 2008].
Pathophysiology
It has long been accepted that the IGEs have a strong genetic component [Berkovic et al., 1998], and given the incidence of JME as a subtype of IGE, much research has been done to understand the underlying genetic basis of JME better. While JME appears to be a relatively homogeneous epilepsy syndrome, there are a number of studies implicating various genetic abnormalities. These include mutations in many different ion channels, including calcium, potassium, and chloride channels linked to at least seven different loci. There are also nonionic mechanisms that relate to neural migration during cortical development, and may explain some of the imaging and postmortem pathologic findings seen in these patients [de Nijs et al., 2009]. For further details about the specific genetic mutations and channels involved, please refer to Chapter 52.
Imaging
Imaging studies in patients with JME have indicated subtle structural changes in mesiofrontal cortex, with an increase in cortical gray matter [Woermann et al., 1999], although this has not been replicated in subsequent studies [Roebling et al., 2009]. These findings are interesting, given that the generalized discharges seen on routine EEGs often have a frontal predominance. This has been further investigated with dense array EEG, which further localized typical 4–6-Hz epileptiform activity to the orbitofrontal/medial frontopolar cortex, whereas some patients also demonstrated basal-medial-temporal sources [Holmes et al., 2010]. These electrophysiologic and anatomic abnormalities appear to be concordant with some of the neuropsychological deficits that are seen in patients with JME. Specific attention has been paid to executive dysfunction that has been demonstrated to correlate with smaller thalamic volumes and more frontal cerebrospinal fluid volumes in children with recent-onset JME when compared to children with benign rolandic epilepsy [Pulsipher et al., 2009].
Treatment
Valproate has long been considered the first-line agent, with reports of up to 86 percent of patients becoming seizure-free for at least 1 year [Penry et al., 1989]. Valproate has also been shown to be particularly effective in those patients with photosensitivity [Covanis et al., 2004]. Despite this efficacy, valproate has many side effects that may lead to significant comorbidities, including weight gain, hyperactivity, transaminitis, thrombocytopenia, pancreatitis. Women of child-bearing age must also consider the possible long-term cognitive effects on a fetus [Meador et al., 2009]. Due to these concerns, many newer AEDs are also commonly used, including lamotrigine, topiramate, zonisamide, and levetiracetam.
Lamotrigine is now accepted to be a broad-spectrum AED that is effective against both partial and generalized seizures [Biton et al., 2005]. Small studies have also shown it to be effective specifically in the management of patients with JME, both as monotherapy and polytherapy, resulting in seizure-free rates between 40 and 83 percent [Buchanan, 1996; Prasad et al., 2003]. LTG is also very well tolerated; however, it does have the potential for exacerbating myoclonic seizures [Prasad et al., 2003; Crespel et al., 2005]. The combination of LTG and VPA has been reported to be particularly effective in both partial and generalized epilepsy, including JME, and suggests the possibility of a synergistic effect [Brodie and Yuen, 1997].
Topiramate is also a broad-spectrum AED that has been shown to be effective in patients with JME, although there are no published studies on its use as monotherapy [Biton and Bourgeois, 2005]. It appears to be equally effective as LTG and VPA when used as polytherapy, although there is a suggestion that tolerability may be inferior [Prasad et al., 2003].
Zonisamide (ZNS) is another broad-spectrum AED with multiple proposed mechanisms of action. Small studies have shown ZNS to be broadly effective against all of the seizure types in JME, with 69, 62, and 38 percent of patients being free of GTC, myoclonic, and absence seizures, respectively [Kothare et al., 2004].
Levetiracetam is gaining a reputation as a broad-spectrum agent, as it has shown to be effective in patients with various forms of idiopathic generalized epilepsies including epilepsy with eyelid myoclonias and absence [Striano et al., 2009], as well as JME [Specchio et al., 2006; Noachtar et al., 2008]. In a study of 120 patients (93 percent of whom had JME) randomized to add-on levetiracetam or placebo, 25 percent were free of myoclonic seizures and 21.7 percent were free of all seizures, including GTCs, during the 12-week treatment phase [Noachtar et al., 2008]. In another open-label study of 48 patients with JME (10 newly diagnosed and 38 resistant to prior treatment), 73 percent were free of GTCs and 37.5 percent were free of myoclonic seizures over the study period, with a mean follow-up of 19 months [Specchio et al., 2006].
Benzodiazepines are widely accepted as having excellent antimyoclonic effects. This is also true of the myoclonic seizures seen in JME, with up to 88 percent of patients on clonazepam having complete control of myoclonic seizures but only 43 percent becoming free of GTCs [Obeid and Panayiotopoulos, 1989]. For this reason, benzodiazepines are not recommended as monotherapy for patients with JME, but can be extremely successful as add-on therapy when myoclonic seizures persist.
Outcomes
It has been widely accepted that JME is a lifelong condition with a high rate of relapse if weaned off AED therapy; however, some recent long-term follow-up studies have shown conflicting results [Martinez-Juarez et al., 2006; Baykan et al., 2008; Camfield and Camfield, 2009]. Furthermore, one must look at specific seizure types, as it appears the natural history of myoclonic seizures decreases after the fourth decade of life, independent of the on-going occurrence of the other seizure types [Baykan et al., 2008].
In contrast, patients in the CAE evolving into JME group fared much worse, with only 3 of 35 individuals achieving complete seizure freedom [Martinez-Juarez et al., 2006]. Although GTCs were controlled with medications in about 66 percent of patients, absence seizures persisted in 63 percent. These data support the conclusion that JME does appear to be a lifelong disease, specifically in the subgroup of patients with CAE evolving into JME. However, there is still a small subgroup (9 percent) of patients in whom the disease may not be lifelong, and based on these data, it is uncertain whether some of the patients who have been seizure-free on monotherapy for many years would be able to come off AED treatment successfully.
The study by Camfied and Camfield seems to suggest that this indeed may be a possibility. In 24 JME patients with a follow-up period of 25.8 years, 4 individuals (17 percent) were free of all seizures and off AEDs. The mean duration of therapy was 6.5 years before discontinuation. Two additional patients attempted to come off AEDs; they relapsed but later were able to be successfully weaned off medication and remain seizure-free. Another three patients (12.5 percent) had myoclonus only [Camfield and Camfield, 2009]. These results suggest that some patients with JME can successfully discontinue medications. Unfortunately, there do not appear to be clinical or EEG variables that can predict which patients will achieve this result. The decision to discontinue AEDs should be individualized for every patient, and a detailed discussion, with specific attention to the frequency of lifestyle provoking factors, is necessary in order to arrive at a safe and well-informed decision.
Progressive Myoclonic Epilepsies
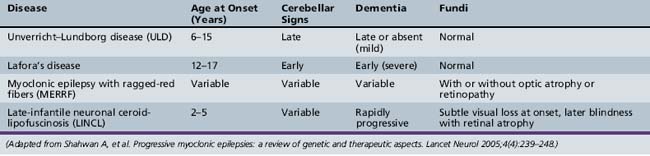
The PME’s are a group of genetically inherited disorders characterized by both epileptic and nonepileptic myoclonus, generalized tonic-clonic seizures, and progressive neurological deterioration, resulting in dementia, ataxia, and various forms of tremor. Recent advances in the genetic basis of these disorders are leading to a better understanding of how each of the disease processes differs. The four most common PMEs will be discussed (Table 56-3).
Unverricht–Lundborg Disease (ULD)
Unverricht–Lundborg disease (ULD) is the most common PME. It usually presents between the ages of 6 and 15 years, with the hallmark presenting symptom being stimulus-sensitive or action myoclonic jerks, which occur in up to half of patients [Lehesjoki, 2002]. Generalized tonic-clonic seizures are also a common feature early in the disease. At initial presentation, neurological examination and EEG studies may be normal, or the EEG may show generalized spike/wave discharges similar to those seen in IGE. It is not until the patient shows signs of progressive ataxia, intention tremor. and dysarthria that one begins to suspect PME clinically. As the disease progresses, the EEG becomes slow, with more frontally predominant polyspike/wave or spike-wave discharges, ranging in frequency between 2 and 6 Hz [Kyllerman et al., 1991]. MRI findings may be normal or may show nonspecific findings, such as cerebellar atrophy and reduced bulk of the basis pontis and medulla [Mascalchi et al., 2002].
The underlying cause of ULD is linked to an abnormality in the cystatin B gene on chromosome 21q22.3 [Lehesjoki et al., 1991]. Although the exact pathophysiology is unknown, mutations in cystatin B appear to lead to accelerated apoptosis, which may explain the progressive neurologic decline [Delgado-Escueta et al., 2001].
The diagnosis of ULD can be made by mutation testing of the cystatin B gene. There is no specific treatment for ULD, but improvements in seizure management with newer-generation AEDs has improved overall prognosis. VPA has been the mainstay of initial treatment; however, recent studies have shown a beneficial effect of levetiracetam, especially if started early [Magaudda et al., 2004].
Lafora’s Disease
Lafora’s disease (LD) has slightly later onset than ULD but has a more rapid decline, with many patients dying within 10 years of onset. Patients share similar clinical characteristics with other PMEs featuring seizures and myoclonus, but have a more prominent dementing component. Seizure types can be varied, but one peculiar type is that of occipital seizures with hallucinations and transient blindness [Minassian, 2001]. All patients are initially normal neurologically, but may have seizures indistinguishable from IGE until progressive neurological decline is noted. EEG findings early in the course may also resemble IGE, including photosensitivity, but the photosensitivity appears to be maximal at low frequencies between 1 and 6 Hz [Kobayashi et al., 1990]. As the disease progresses, the EEG background becomes slow and disorganized, and there is an evolution of the spike/wave discharges, the 3 Hz discharges becoming faster 6–12 Hz ones [Yen et al., 1991].
The underlying cause of LD is linked to two different sites on chromosome 6 – 6q24 and 6p22 – coding for two different genes: EPM2A (larforin) and NHLRC1 respectively [Minassian et al., 1998; Chan et al., 2003]. The former appears to be involved in regulation of protein folding and the latter may play a role in dendritic transport in neurons. How dysfunction in each of these functions leads to the disease remains unclear.
The diagnosis of LD can be made by testing for mutations in the EPM2A gene that accounts for 80 percent of cases [Minassian et al., 1998]. In those cases where mutation testing is negative but the clinical phenotype is giving cause for concern, Lafora bodies can be detected in skin biopsy specimens. Treatment remains palliative.
Myoclonic Epilepsy with Ragged-Red Fibers (MERRF)
Myoclonic epilepsy with ragged-red fibers (MERRF) is a mitochondrial disease characterized by myoclonic epilepsy, ataxia, and ragged-red fibers on muscle biopsy. Multiple other clinical signs commonly seen in mitochondrial disorders may also be seen, such as myopathy, hearing loss, short stature, neuropathy, and optic atrophy [Chinnery et al., 1997]. EEG findings early in the disease are similar to the other PMEs, with 2–5-Hz generalized spike-wave discharges, and gradual background slowing as the disease progresses [So et al., 1989]. Imaging findings include basal ganglia calcifications and brain atrophy [DiMauro et al., 2002]. The key to differentiating MERFF from other PMEs is the constellation of other peripheral nervous system findings.
Diagnosis is made by genetic testing for the most common mutation in the tRNA (MTTK) gene of the mitochondrial DNA, which is seen in 90 percent of typical patients [Shoffner et al., 1990]. There is no specific treatment for MERRF although various antioxidants are commonly prescribed, as is customary in other mitochondrial disorders. VPA can be used, although l-carnitine supplementation is recommended [Tein et al., 1993].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


