Figure 13.1 Facioscapulohumeral dystrophy
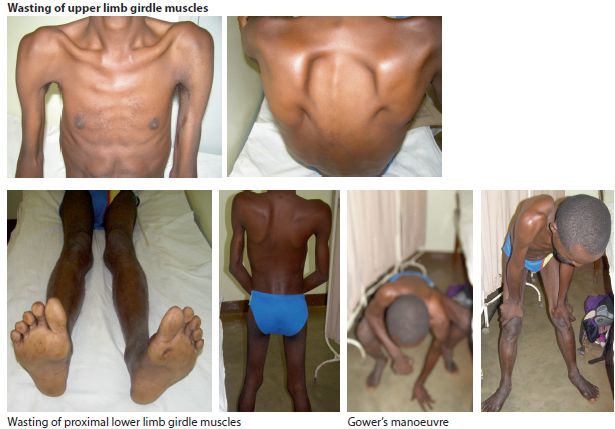
Figure 13.2 Limb girdle muscular dystrophy
Key points
INFLAMMATORY MYOPATHIES
DERMATOMYOSITIS AND POLYMYOSITIS
Dermatomyositis and polymyositis are the main inflammatory myopathies. They are defined by inflammation in muscles and their general characteristics are presented below (Table 13.2). Their approximate frequency in high income countries is 5/100,000. In Africa they mostly affect the age group 20-40 yrs but occur in other age groups. Females are more commonly affected than males.
Table 13.2 Characteristics of inflammatory myopathies
| Characteristics | Polymyositis | Dermatomyositis |
| age group | 20-40 yrs | 10-50 yrs |
| female:male ratio | 2:1 | 2:1 |
| onset | months | weeks to months |
| proximal muscle weakness | yes | yes |
| skin involvement | no | yes |
| malignancy | no | slight risk |
| elevation in ESR & CK | marked | marked |
| response to steroids | yes | yes |
Clinical features
Patients present with sub acute mainly proximal muscle weakness and pain over months but occasionally over weeks. Dysphagia may occur because of involvement of pharyngeal muscles. In dermatomyositis both skin and muscle are involved. On examination, there is marked symmetrical proximal weakness and signs of muscle atrophy in long standing disease. There is a characteristic blue-purple rash, plus oedema of upper eye lids (heliotrope), erythema over cheeks, knuckles and chest (Fig. 13.3) and dilated capillaries at the base of finger nails.
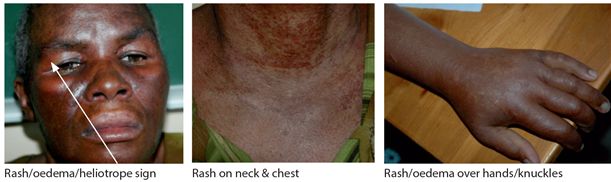
Figure 13.3 Dermatomyositis
Investigations
The ESR and serum CK are markedly elevated in polymyositis. The diagnosis is confirmed by muscle biopsy. Malignancy should be screened for in patients with dermatomyositis.
Management
The management of polymyositis is based on corticosteroids and immunosuppressant drugs. Treatment is with prednisolone 1mg/kg or 60 mg/po/daily. After months, when CK returns to normal and there is clinical improvement, the steroids can be reduced gradually by 5 mg decrements per dose on the alternate day, until the steroids are being administered only on alternate days. If the improvement is maintained, the prednisolone can be further decreased by 10 mg decrements every 4 weeks, until a maintenance dose is found. The usual maintenance dose is 5-10 mg prednisolone on alternate days. This may need to be continued for a further period of 3-6 months. Consider using an immunosuppressant drug from the start with steroids. The drug of choice is azathioprine 2.5 mg/kg/day. Start with 50 mg per day and increase by weekly intervals to 125-150 mg daily in divided doses. The main side effects are bone marrow suppression and hepatic toxicity. A FBC and liver function should be checked at the start and at least every four months during treatment with azathioprine. Alternatives are methotrexate or cyclophosphamide.
Key points
INCLUSION BODY MYOSITIS
Inclusion body myositis (IBM) is another form of progressive inflammatory muscle disease which occurs in people >50 yrs. It is relatively common in high income countries but its frequency in Africa is not known. It occurs more frequently in older males and presents with painless proximal weakness with selective involvement of finger flexors and quadriceps muscles and frequently involves swallowing. Diagnosis is established by muscle biopsy and long term treatment is unsatisfactory. The differential diagnosis of inflammatory myopathies includes myasthenia gravis, non inflammatory myopathies and neuropathies.
OTHER MYOPATHIES
There is a large group of acquired non inflammatory myopathies. The main causes are outlined in Table 13.3. Patients may range from being relatively asymptomatic with just muscle wasting to having severe weakness of limbs and trunk muscles. Most cases remit once the underlying disorder is treated or the offending drug is withdrawn.
Table 13.3 Causes of acquired non inflammatory myopathies
| Classification | Causes |
| infections | HIV, TB |
| endocrine | thyroid disorders Cushing’s disease |
| neoplastic | any malignancy |
| metabolic | hyper/hypokalaemia |
| organ failure | chronic cardiac, respiratory, liver disease |
| drugs | alcohol, steroids, statins, ARTs (zidovudine) |
MYASTHENIA GRAVIS
Myasthenia gravis (MG) is an uncommon autoimmune disease that is caused by acetylcholine receptor antibodies (AChRA) at the neuromuscular junction. The antibody binds to the post synaptic acetylcholine receptor sites which makes them unavailable for the transmission of nerve impulses. It is a worldwide disorder with an incidence in high income countries of around 3-30/1,000,000 per year and with most cases expected to survive, the approximate prevalence rate is much higher at around 10/100,000 with a reported increase over time in age related MG. The incidence rates across Africa are not known, but a detailed study from Cape Town in SA using AChRAs as in indicator of MG reported an incidence rate there of 12.6/1,000,000 which is similar to that in high income countries. MG affects all age groups but mainly females in the age group 20-40 yrs and males in an older age group. It is associated with hyperplasia of the thymus (70%) and less commonly with thymoma (10%).
Clinical features
Symptoms
The most important diagnostic feature of MG is fatigable muscle weakness. In the early stages, weakness may be transient and variable. The busiest muscles at rest are the ones most commonly affected and patients frequently present for the first time with involvement of the extra ocular muscles (diplopia), eyelid (ptosis) and bulbar muscles (dysphagia) (Fig. 13.4). These may be accompanied by proximal weakness of the limbs and involvement of the face, neck, and trunk; typically the weakness worsens after exercise or at the end of the day.
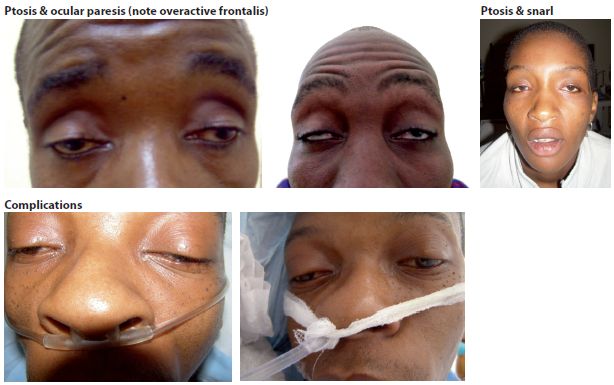
Figure 13.4 Myasthenia gravis
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


