36 Myotonic Muscle Disorders and Periodic Paralysis Syndromes
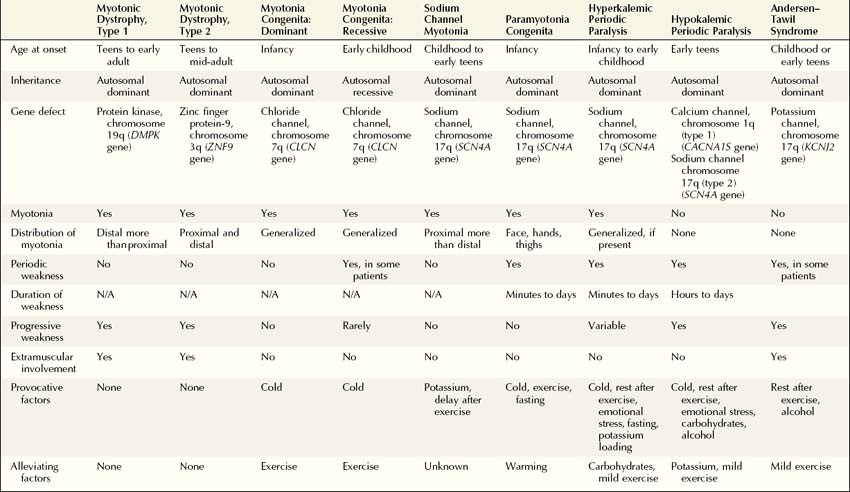
The myotonic muscle disorders and periodic paralysis syndromes compose a group of disorders characterized by muscle stiffness, pain, and sometimes weakness, which may be intermittent or constant. Evaluation of these disorders in the electromyography (EMG) laboratory is particularly gratifying, as the EMG accompaniment of myotonia is easily recognized by the experienced electromyographer. Clinically, myotonia is characterized by delayed muscle contraction after activation. Myotonia can also be demonstrated after percussion of the muscle. On EMG, myotonic discharges produce a distinctive revving engine sound. This results from the spontaneous firing of muscle fibers that wax and wane in frequency and amplitude, producing this unmistakable sound (Figure 36–1). The myotonic potential may take the form of either a positive wave or a brief spike potential, thus identifying the source generator as a muscle fiber. Myotonia can be induced by mechanical stimulation, such as percussion of the muscle or movement of the EMG needle, or may follow voluntary muscle contraction. Clinically, myotonia is noted most frequently in the myotonic muscle disorders and in some of the periodic paralysis syndromes (Box 36–1). Patients describe an inability to relax their muscles after contraction, such as during hand grip. In addition, myotonia may be experienced by the patient as muscle stiffness.

Box 36–1
Classification of Myotonic and Periodic Paralysis Disorders
I. Inherited myotonic muscle/periodic paralysis disorders
II. Acquired periodic paralysis disorders
III. Muscle disorders associated with electromyographic myotonia
IV. Drugs that unmask or precipitate myotonia either clinically or on electromyographic examination
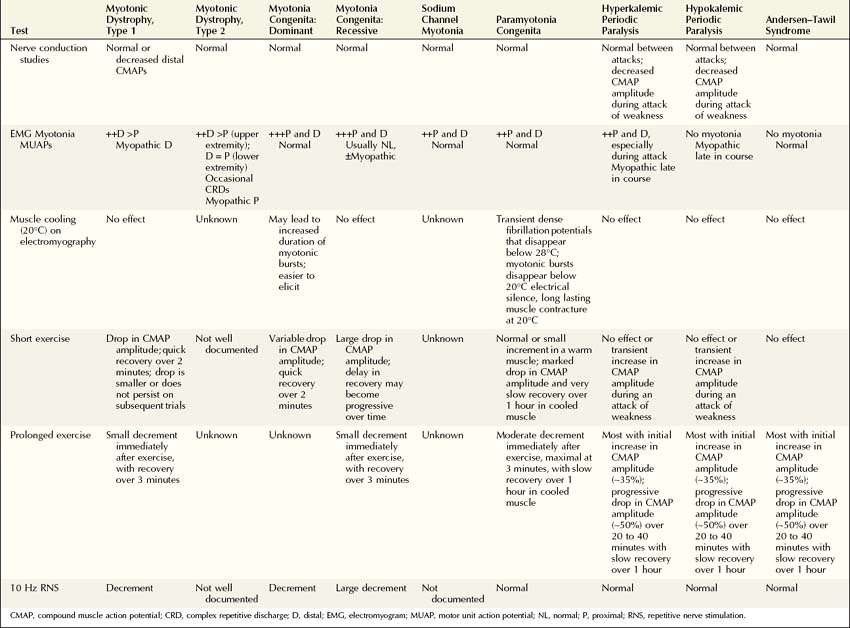
Genetic linkage and mutational analyses have identified the molecular basis for several of the myotonic muscle disorders and periodic paralysis syndromes, resulting in the classification of these disorders based on a specific ion channel or protein kinase defect. Classification of these disorders now can be accomplished based on clinical, electrophysiologic, and molecular findings (Table 36–1). Once myotonia is identified on clinical or needle EMG examination, the electrophysiologic evaluation is directed toward answering several key questions to arrive at the correct diagnosis. To answer these questions, a variety of tests can be performed in the EMG laboratory to distinguish among the dystrophic and nondystrophic myotonic muscle disorders, the periodic paralysis syndromes, and other disorders of muscle with accompanying EMG myotonia. In addition to routine nerve conduction studies and needle EMG, muscle cooling, exercise testing, and repetitive nerve stimulation often are very helpful in differentiating among these disorders (Box 36–2).
Box 36–2
Protocol for Evaluation of Myotonic and Periodic Paralysis Disorders
1. Routine motor and sensory nerve conduction studies should be done first. Generally one or two motor and sensory conduction studies and corresponding F responses in an upper and lower extremity should be performed. Distal CMAPs may be low in the dystrophic myopathies. Proceed to needle EMG.
2. Needle EMG study is carried out after standard conduction studies are completed. The study should include proximal and distal muscles of one upper and lower extremity, as well as facial and paraspinal muscles. Careful note should be made of abnormal spontaneous activity, including myotonic discharges, complex repetitive discharges, fibrillation potentials and positive waves, and MUAP potential configuration and recruitment pattern.
3. Muscle cooling is carried out if there is a clinical suspicion of paramyotonia congenita.
4. Short exercise test is performed if steps 1, 2, and 3 do not yield a definitive diagnosis.
5. Prolonged exercise test is performed if steps 1, 2, 3, and 4 do not yield a definitive diagnosis.
CMAP, compound muscle action potential; EMG, electromyography; MUAP, motor unit action potential.
Exercise Testing
Short Exercise Test
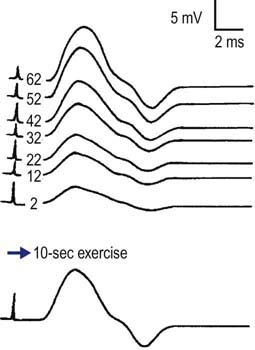
For the short exercise test, the patient is asked to rest for about 5 minutes while a CMAP is recorded every minute, to ensure that the baseline is stable and does not decrease at rest. The patient is then asked to perform maximal voluntary contraction for 5 to 10 seconds. Immediately afterward, a CMAP is recorded. If a decrement in amplitude is seen, then a CMAP is recorded every 10 seconds until the CMAP recovers to baseline (typically 1–2 minutes) (Figure 36–2). If a decrement occurs after brief exercise and then recovers, the same procedure is repeated several times to see if the decrement continues to occur or habituates, which can help differentiate among some of the myotonic syndromes (discussed later).
Prolonged Exercise Test
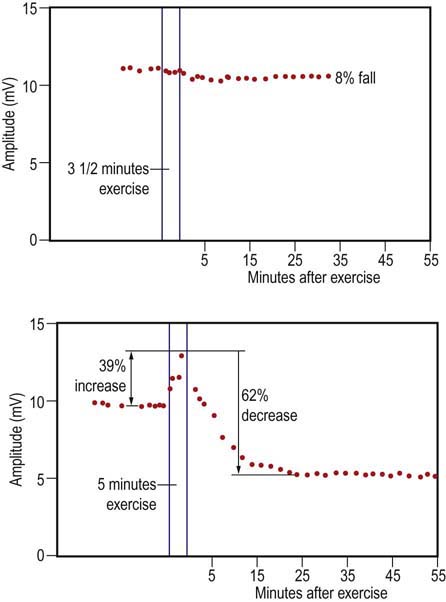
For the prolonged exercise test, the recording procedure is the same. The patient is asked to rest for about 5 minutes while a CMAP is recorded every minute, to ensure the baseline is stable and does not decrease at rest. After ensuring a stable baseline, the patient is asked to voluntarily contract his or her muscle maximally for 5 minutes, resting every 15 seconds for a few seconds. After the 5 minutes of exercise are complete, the patient relaxes completely. A CMAP is recorded immediately and then every 1 to 2 minutes for the next 40 to 60 minutes. In the periodic paralysis syndromes, both inherited and acquired, the CMAP amplitude may be unchanged or slightly larger immediately after prolonged exercise and then decline substantially over the next 20 to 60 minutes (Figure 36–3).
Repetitive Nerve Stimulation
When all the available electrophysiologic techniques are used, the correct diagnosis usually can be determined by answering several key questions (Table 36–2):
1. Are routine nerve conduction studies normal?
3. Is there an effect of muscle cooling on the needle examination?
4. What does the short exercise testing show?
5. What does the prolonged exercise testing show?
Dystrophic Myotonic Muscle Disorders
Myotonic Dystrophy
Myotonic Dystrophy Type 1
Clinical
There is a distinctive clinical appearance characterized by bifacial weakness, temporal wasting, and frontal balding, resulting in a narrow, elongated face and horizontal smile, with ptosis, and distal muscle wasting and weakness (Figure 36–4). Patients with a smaller CTG repeat may not have the typical facial appearance. Weakness of neck flexion is also an early sign, and patients may notice difficulty lifting their head off the pillow or a tendency for the head to fall backwards during acceleration. DM1 is distinguished from many of the other myotonic disorders by the progressive distal weakness as well as involvement of several organ systems outside of skeletal muscle resulting in cataracts, cardiac conduction and pulmonary defects, endocrine dysfunction, testicular atrophy, hypersomnia, gynecologic problems, and, in some patients, mild to moderate cognitive impairment. As in the other myotonic and periodic paralysis syndromes, patients with myotonic dystrophy should be warned against potential anesthetic complications of succinylcholine and anticholinesterase agents.
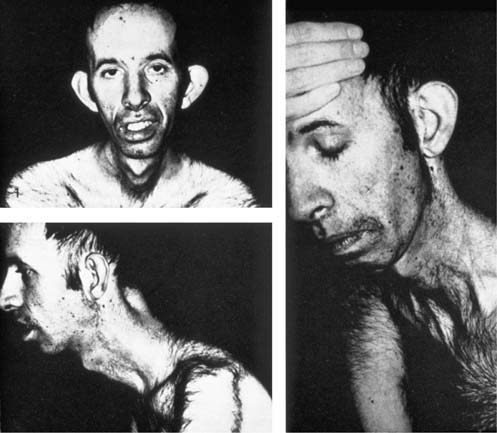
FIGURE 36–4 Typical facies in myotonic dystrophy.
Note frontal balding, ptosis, temporal wasting, elongated face, horizontal smile.
(Reprinted from Brooke, M.H., 1986. A clinician’s view of neuromuscular disease. Williams & Wilkins, Baltimore, with permission.)
Electrophysiologic Evaluation
The electrophysiologic evaluation of DM1 (Table 36–2) consists of routine nerve conduction studies, electromyography, muscle cooling, and exercise testing.
1. Routine motor and sensory nerve conduction studies are normal as a rule. Generally, one motor and sensory nerve conduction study and F responses in an upper and lower extremity will suffice. A mild neuropathy has been described, perhaps secondary to the accompanying endocrine changes. Low CMAP amplitudes may be noted secondary to the distal myopathy in patients with severe disease.
2. Concentric needle EMG of at least one upper and one lower extremity should be performed, in addition to sampling facial and paraspinal muscles. Most but not all patients with DM1 will demonstrate myotonic discharges on EMG. In very mild cases (e.g., in patients with a small increase in the number of repeats), myotonic discharges may be difficult to find. Otherwise, myotonic discharges are generally most prominent in the distal hand, forearm extensor, foot dorsiflexor (tibialis anterior), and facial muscles but usually are not found in proximal muscles. The distribution of myotonic discharges follows the same pattern as the weakness. Myotonic discharges in DM1 are the classic waxing and waning muscle fiber action potentials (Figure 36–5A). MUAP analysis may be difficult because of the myotonic discharges provoked by needle insertion or muscle contraction. However, careful examination reveals myopathic (low amplitude, short duration, polyphasic) MUAPs with early recruitment, which are generally noted in the forearm extensor and tibialis anterior muscles, consistent with the distal predominant weakness on clinical examination.
3. Muscle cooling to 20°C has no appreciable effect on the EMG examination.
4. The short exercise test produces a drop in the CMAP amplitude immediately after exercise. If the CMAP then is recorded every 10 seconds up to 2 minutes, it recovers to baseline. If short exercise is repeated, the decremental response habituates after one or two cycles, with no further decrement in the CMAP occurring immediately after exercise.
5. Repetitive nerve stimulation at 10 Hz produces a decrement, similar to the short exercise test.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


