Chapter 65 Neurocutaneous Syndromes
Tuberous Sclerosis
Tuberous sclerosis complex (TSC) is a disorder of cellular differentiation and proliferation that can affect the brain, skin, kidneys, heart, and other organs. Many clinical features of TSC result from hamartomas, but true neoplasms also occur, particularly in the kidney and brain. Abnormal neuronal migration plays a major additional role in neurological dysfunction (Roach and Sparagana, 2004).
Population-based studies suggest a prevalence of 1 per 6000 to 9000 individuals. However, because of the striking variability of clinical expression, establishing the diagnosis of TSC can be difficult in individuals with subtle findings, and the true prevalence may be considerably higher. Cutaneous findings are usually the first clue that a patient has TSC, but other features may lead to the diagnosis (Box 65.1). In infants, cardiac involvement and seizures frequently are presenting signs, whereas dermatological, pulmonary, or renal involvement may lead to diagnosis in older individuals.
Box 65.1
Diagnostic Criteria for Tuberous Sclerosis Complex
Major Features
Minor Features
1. Multiple randomly distributed pits in dental enamel
2. Hamartomatous rectal polyps‡
4. Cerebral white-matter radial migration lines*,†,i
Definite tuberous sclerosis complex: either two major features or one major feature plus two minor features.
Probable tuberous sclerosis complex: one major plus one minor feature.
Possible tuberous sclerosis complex: either one major feature or two or more minor features.
Reprinted with permission from Roach, E.S., Gomez, M.R., Northrup, H., 1998. Tuberous sclerosis consensus conference: revised clinical diagnostic criteria. J Child Neurol 13, 624-628.
* When cerebral cortical dysplasias and cerebral white-matter migration tracts occur together, they should be counted as one rather than two features of tuberous sclerosis.
† When both lymphangiomyomatosis and renal angiomyolipomas are present, other features of tuberous sclerosis should be present before a definite diagnosis is assigned.
‡ Histological confirmation is suggested.
§ Radiographical confirmation is sufficient.
i One panel member felt strongly that three or more radial migration lines should constitute a major sign.
The inheritance of TSC is as an autosomal dominant trait with variable penetrance. The estimated spontaneous mutation rate for TSC varies from 66% to 86%, depending in part on the completeness of investigation of the extended family. Two genes are responsible for TSC: TSC1, coding for hamartin at chromosome 9q34.3; and TSC2, coding for tuberin adjacent to the gene for adult polycystic kidney disease at chromosome 16p13.3. The clinical features of TSC1 and TSC2 overlap, since the two gene products form a single functional unit that is an upstream modulator in the mTOR (mammalian target of rapamycin) signaling pathway. Both gene products down-regulate small G-protein Ras-homologue enriched in brain (RHEB) activity in this pathway. However, genotype-phenotype studies indicate that individuals with a TSC2 mutation tend to have more severe disease, and the frequency of TSC2 mutations is greater among individuals with spontaneous mutations (Sancak et al., 2005). Multiple mutation types exist in different regions of each gene, and even individuals with identical genetic mutations can have different phenotypes. Molecular diagnostic testing—including prenatal testing—has been available since the early 2000s, and a disease-causing mutation is identified in 85% meeting clinical criteria. Individuals with no mutation identified may have distinctive clinical features. Large genomic deletions and rearrangements are more common in the TSC2 gene compared to TSC1, and more mutations have been identified for TSC2 versus TSC1. TSC2 mutations appear more commonly than TSC1 in patients with subependymal nodules, mental retardation, renal angiomyolipomas, and retinal phakomas. Mental retardation and other neuropsychiatric involvement are more likely in individuals with TSC2 versus TSC1 mutation (Au et al., 2007).
Cutaneous Features
The cutaneous lesions of TSC include hypomelanotic macules, the shagreen patch, ungual fibromas, and facial angiofibromas. Hypomelanotic macules (ash leaf spots) occur in up to 90% of affected individuals (Fig. 65.1). The lesions usually are present at birth but may be seen in the newborn only with an ultraviolet light. Other pigmentary abnormalities include confetti lesions (areas with stippled hypopigmentation, typically on the extremities) and poliosis (a white patch or forelock) of the scalp, hair, or eyelids (Fig. 65.2, D). Hypomelanotic macules are common in normal individuals (Table 65.1), but three or more hypomelanotic papules is a major diagnostic criterion for TSC.
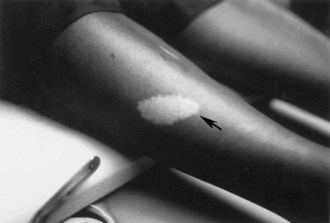
Fig. 65.1 A hypomelanotic macule (ash leaf spot) (arrow) from the leg of a patient with tuberous sclerosis.
(Reprinted with permission from Weiner, D.M., Ewalt, D.H., Roach, E.S., et al., 1998. The tuberous sclerosis complex: a comprehensive review. J Am Coll Surg 187, 548-561.)
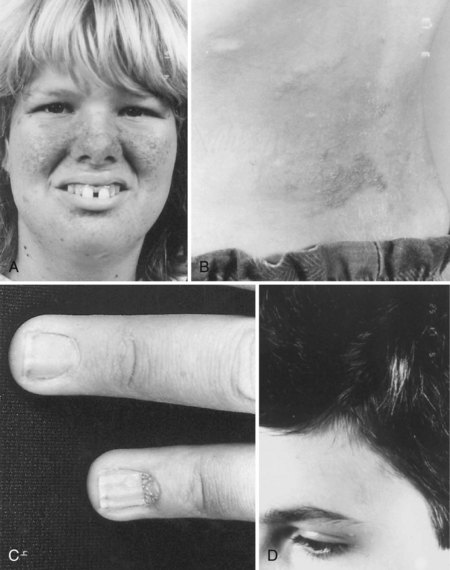
Table 65.1 Frequency of Lesions in Individuals with Tuberous Sclerosis Versus Other Individuals
| Lesion | Tuberous Sclerosis Complex | Other Individuals |
|---|---|---|
| Hypomelanotic macules | Occur in over 95% of tuberous sclerosis (TSC) patients, often with many lesions11 | Occur in up to 5% of the population (but usually fewer than three lesions per person)12 |
| Facial angiofibromas | Eventually seen in 75% but less often in children11 | Seen in individuals with multiple endocrine neoplasia type 1 and in a few sporadic families13 |
| Shagreen patch | Up to 48%11 | Occasional |
| Ungual fibromas | Seen in 15% but often not until develop in adulthood11 | Occasionally sporadic or after nail trauma (but typically one lesion)14 |
| Rhabdomyomas | One or more tumors seen in 47%-65% but much more common below 2 years Up to 51% of patients with rhabdomyomas have TSC15-17 | In 14%-49% of rhabdomyoma patients, there are no other signs of TSC16 |
| Renal angiomyolipoma (AML) | Often multiple AML occur in up to 80% of TSC patients by age 1018 | Sporadic AML occur but are typically solitary |
| Renal cysts | Polycystic kidneys occur in 3%-5% of TSC patients Smaller numbers of renal cysts are present in 15%-20%18 | There are both dominant and recessive polycystic kidney diseases A few cysts are frequent sporadic findings in adults |
| Cortical dysplasia/tubers | 90%-95% and usually multiple lesions are present (magnetic resonance imaging yields highest detection rate)25 | Sporadic cortical dysplasia (typically 1 lesion) is common among individuals who have epilepsy not due to TSC |
| Subependymal nodules | 83%-93%2,25 | Rare, especially if calcified |
| Subependymal giant cell tumors | Up to 15% (using radiographic criteria)19 | Rare in the absence of TSC |
From Roach, E.S., Sparagana, S.P., 2010. Diagnostic criteria for tuberous sclerosis complex. In: Kwiatkowski, D.J., Whittemore, V.H., Thiele, E.A. (Eds.), Tuberous Sclerosis Complex: Genes, Clinical Features, and Therapeutics. Weinheim, Wiley-VCH Verlag GmbH & Co., pp. 21-25. Used with permission.
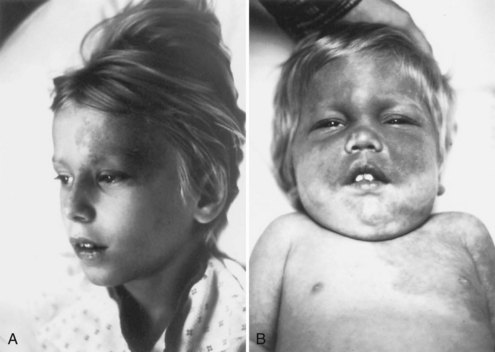
Facial angiofibromas (adenoma sebaceum) consist of vascular and connective tissue elements. Although considered specific for TSC, they are found in only three-fourths of affected individuals and often appear several years after other means have established the diagnosis. The lesions typically become apparent during the preschool years as a few small red macules on the malar region; they gradually become papular, larger, and more numerous, sometimes extending down the nasolabial folds or onto the chin (see Fig. 65.2, A). Laser therapy may be a useful therapeutic intervention and particularly helpful in early childhood or just prior to puberty, before a time when growth may be rapid and more aggressive interventions warranted.
The shagreen patch most often is found on the back or flank area; it is an irregularly shaped, slightly raised, or textured skin lesion (see Fig. 65.2, B). Only 20% to 30% of patients with TSC have one patch, which may not be seen in young children. Usually considered specific for TSC, ungual fibromas are nodular or fleshy lesions that arise adjacent to (periungual) or underneath (subungual) the nails (see Fig. 65.2, C). These can occur as a single lesion after trauma in normal individuals. Ungual fibromas occur in only 15% to 20% of patients with TSC, more likely in adolescents or adults.
Neurological Features
Most patients with mental retardation have epilepsy, but many have seizures and normal intelligence. The number of subependymal lesions does not correlate with the clinical severity of TSC, but patients with MRI evidence of numerous cortical lesions tend to have more cognitive impairment and more difficulty with seizure control. The most abnormal regions seen on MRI tend to coincide with focal abnormalities of the electroencephalogram (EEG). The likelihood of intellectual disability in patients with TSC probably is overestimated, and the severity of intellectual dysfunction ranges from borderline to profound mental retardation. However, in addition to intellectual disability, many children with TSC have serious behavioral disorders. Autistic behavior, hyperkinesis, aggressiveness, and frank psychosis sometimes occur, either as isolated problems or in combination with epilepsy or intellectual deficit. The prevalence of autistic spectrum disorders is 25% to 50% and equal between boys and girls. Behavioral problems are frequent and independent of intellectual ability (de Vries, 2010). Mood disorders also are increased.
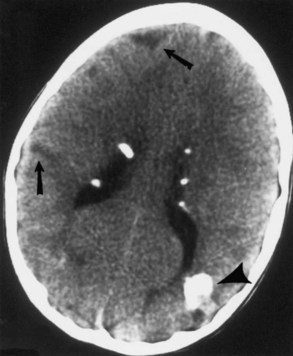
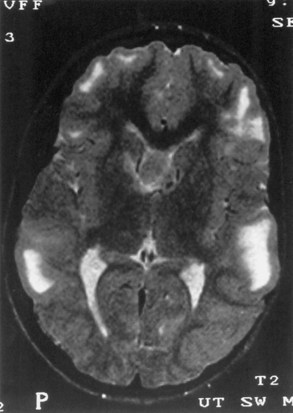
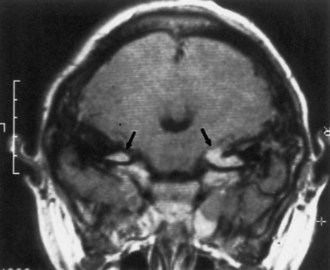
Computed tomography (CT) best demonstrates the calcified subependymal nodules that characterize TSC (Fig. 65.3). CT sometimes shows superficial cerebral lesions, but they are far more obvious with T2-weighted MRI (Fig. 65.4). T2-weighted scans show evidence of abnormal neuronal migration in some patients as high-signal linear lesions running perpendicular to the cortex. SENs along the ventricular surface give the characteristic appearance of candle guttering. More than one-fourth of patients with TSC show cerebellar anomalies.
Subependymal giant-cell astrocytomas (SEGAs) develop in 6% to 14% of patients with TSC. Unlike the more common cortical tubers and SENs, giant-cell astrocytomas can enlarge (Fig. 65.5) and cause symptoms of increased intracranial pressure, particularly if extension into the lateral ventricles creates an obstructive hydrocephalus. Clinical features include new focal neurological deficits, increased intracranial pressure, unexplained behavior change, or deterioration of seizure control. Acute or subacute onset of neurological dysfunction may result from sudden obstruction of the ventricular system by an intraventricular SEGA. Rarely, acute deterioration occurs because of hemorrhage into the tumor itself.
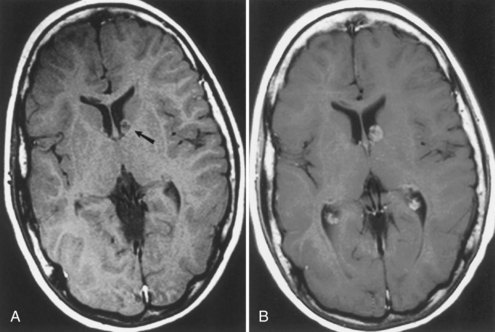
Giant-cell tumors are usually benign but locally invasive, and early surgery can be curative. Identification of an enlarging SEGA before the onset of symptoms of increased intracranial pressure or appearance of new neurological deficits is ideal. Periodic screening to identify SEGA may improve surgical outcome. Recent work suggests that rapamycin inhibits the growth of SEGAs (Franz et al., 2006).
Retinal Features
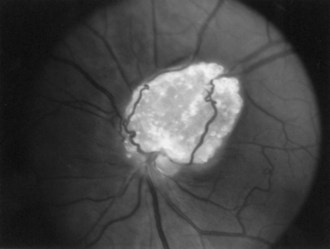
The frequency of retinal hamartomas in TSC varies from almost negligible to 87% of patients, probably reflecting the expertise and technique of the examiner. Pupillary dilatation and indirect ophthalmoscopy are important, particularly in uncooperative children. Findings vary from classic mulberry lesions adjacent to the optic disc (Fig. 65.6) to plaque-like hamartoma or depigmented retinal lesions. Most retinal lesions are clinically insignificant, but some patients have visual impairment caused by large macular lesions, and very few patients have visual loss caused by retinal detachment, vitreous hemorrhage, or hamartoma enlargement. Occasionally, patients have a pigmentary defect of the iris. Funduscopic examination is valuable at the time of diagnosis, to monitor existing abnormalities or evaluate for new symptoms. The histological features of retinal hamartomas, subependymal nodules, and subependymal giant-cell astrocytomas are similar.
Systemic Features
Cardiac
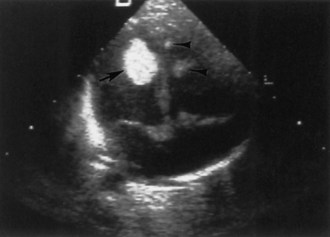
Approximately two-thirds of patients with TSC have a cardiac rhabdomyoma, but few demonstrate clinical symptoms. Cardiac rhabdomyomas are hamartomas, tend to be multiple, and involute with time. These lesions sometimes are evident on prenatal ultrasound testing (Fig. 65.7)—usually after 24 weeks gestational age—and most who develop cardiac dysfunction present soon after birth with heart failure. A few children later develop cardiac arrhythmias or cerebral thromboembolism from the rhabdomyomas. The cause of congestive heart failure is either by obstruction of blood flow by intraluminal tumor or by lack of sufficient normal myocardium to maintain perfusion. Some patients stabilize after medical treatment with digoxin and diuretics and eventually improve; others require surgery. Echocardiography and electrocardiogram establish the diagnosis. Arterial aneurysms can occur. For existing rhabdomyomas, perform surveillance studies every 6 to 12 months until stabilization or involution occurs. The size of these lesions may increase with hormone exposure—a consideration in the neonate, pubertal individual, and child treated with ACTH for infantile spasms.
Renal
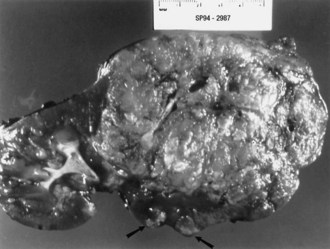
Renal angiomyolipomas occur in up to three-fourths of patients with TSC, usually presenting by 10 years of age. Most of these lesions are histologically benign tumors with varying amounts of vascular tissue, fat, and smooth muscle (Fig. 65.8). Bilateral tumors or multiple tumors in a kidney are common. The prevalence and size of renal tumors increase with age, and tumors larger than 4 cm are much more likely to become symptomatic than smaller tumors. Renal cell carcinoma or other malignancies are less common but affect TSC patient at younger ages than the general population. Coalescing angiomyolipomata can contribute to end-stage renal disease. Endovascular embolization of the larger renal angiomyolipomata prevents hemorrhage and other complications (Ewalt et al., 2005). Rapamycin limits the growth of these tumors, at least transiently.
Neurofibromatosis
A mutation of the NF2 gene on chromosome 22 causes NF2. The NF2 protein product is schwannomin or merlin. The NF2 gene suppresses tumor function. Dysfunction of the NF2 gene accounts for the occurrence of multiple central nervous system (CNS) tumors in patients with NF2. The NF2 gene has several different mutations. The clinical severity may be related to the nature of the NF2 mutation; missense mutations that allow some protein function tend to produce milder clinical forms, whereas frameshift and nonsense mutations that produce stop codons preventing the production of any protein often cause severe disease (Evans, 2004).
If several characteristics are present, the diagnosis of either NF1 or NF2 is obvious, especially when another family member is affected. The diagnosis is difficult when the clinical features are atypical and the family history is negative. Very young children may have fewer apparent lesions, making definitive diagnosis difficult. Diagnostic criteria (Box 65.2) help to resolve some of these questionable cases, but specific gene testing is replacing the use of clinical criteria. Screening for the NF1 gene is technically difficult because the gene is large and several different mutations are causative. Commercially available studies have a 30% false-negative rate. Some suggest the diagnosis of NF2 based on multiple meningiomas or nonvestibular schwannomas even without family history or classic bilateral vestibular schwannomas.
Box 65.2
Diagnostic Criteria for Neurofibromatosis
Neurofibromatosis Type 1 (Any Two or More)
Six or more café-au-lait lesions more than 5 mm in diameter before puberty and more than 15 mm in diameter afterward
Freckling in the axillary or inguinal areas
Two or more neurofibromas or one plexiform neurofibroma
A first-degree relative with neurofibromatosis type 1
A characteristic bony lesion (sphenoid dysplasia, thinning of the cortex of long bones, with or without pseudoarthrosis)
Neurofibromatosis Type 2
Bilateral eighth nerve tumor (shown by magnetic resonance imaging, computed tomography, or histological confirmation)
A first-degree relative with neurofibromatosis type 2 and a unilateral eighth nerve tumor
A first-degree relative with neurofibromatosis type 2 and any two of the following lesions: neurofibroma, meningioma, schwannoma, glioma, or juvenile posterior subcapsular lenticular opacity
Data from Neurofibromatosis. Conference statement, 1988. National Institutes of Health Consensus Development Conference. Arch Neurol 45, 575-578.
Cutaneous Features of Neurofibromatosis Type 1
Cutaneous lesions of NF1 (Fig. 65.9) include café-au-lait spots, subcutaneous neurofibromas, plexiform neurofibromas, and axillary freckling. Café-au-lait spots are flat, light to medium brown areas that vary in shape and size. They typically are present at birth but increase in size and number during the first few years of life. Later in childhood, skin freckling, 1 to 3 mm in diameter, often occurs symmetrically in the axillae (Crowe sign) and other intertriginous regions. Most children with 6 or more café au lait spots as their only diagnostic criterion will go on to meet diagnostic criteria, usually by age 6.
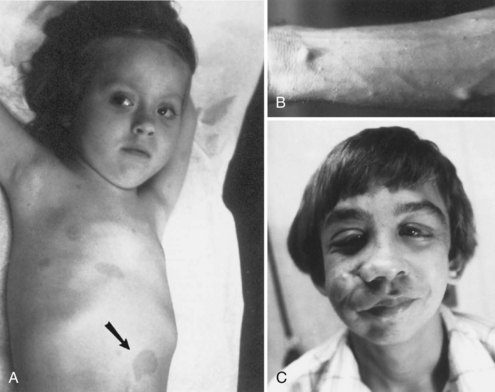
Neurofibromas are benign tumors arising from peripheral nerves (see Fig. 65.9, B). These tumors are composed predominantly of Schwann cells and fibroblasts but contain endothelial, pericytes, and mast cell components. Neurofibromas can develop at any time; their size and number often increase after puberty.
Plexiform neurofibromas often occur on the face and can cause substantial deformity (see Fig. 65.9, C). Patients with plexiform tumors of the head, face, or neck and those who presented before 10 years of age are more likely to do poorly (Needle et al., 1997). Plexiform neurofibromas have a 5% to13% lifetime risk of malignant degeneration into malignant peripheral nerve sheath tumors. Malignant peripheral nerve sheath tumors (MPNST) carry poor 5-year survival rates despite treatment with surgery, chemotherapy, and radiation. PNFs and MPNST are difficult to distinguish radiographically and sometimes even pathologically.
Systemic Features of Neurofibromatosis Type 1
Lisch nodules are pigmented iris hamartomas (Fig. 65.10). They are pathognomonic for NF1. Lisch nodules do not cause symptoms; their significance lies in their implications for the diagnosis of NF1. Lisch nodules are often not apparent during early childhood, so their absence does not exclude the diagnosis of NF1. Rarely, children with NF1 have retinal hamartomas, but these usually remain asymptomatic.
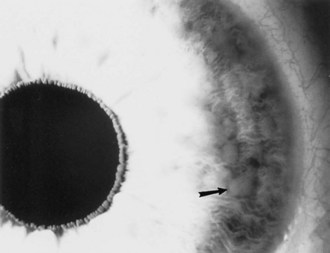
Fig. 65.10 Lisch nodules (arrow) of the iris in a patient with neurofibromatosis type 1.
(Reprinted with permission from Roach, E.S., 1992. Neurocutaneous syndromes. Pediatr Clin North Am 39, 591-620.)
Neurological Features in Neurofibromatosis Type 1
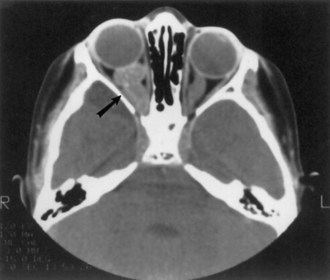
Optic nerve glioma (Fig. 65.11) is the most common CNS tumor caused by NF1. Approximately 15% of patients with NF1 have unilateral or bilateral optic glioma. The growth rate of these tumors varies, but they tend to behave less aggressively in patients with NF1 than those without NF1. When symptomatic, the presenting features are optic atrophy, progressive vision loss, pain, or proptosis. Precocious puberty is a common presenting feature of chiasmatic optic nerve tumors in children with NF1. Management options include observation with serial brain MRI or treatment with radiation, chemotherapy, or excision. Radiation is less favored, especially given possible exacerbation of vasculopathy in this population.
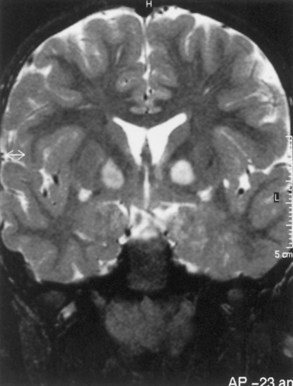
Macrocephaly is seen in half of NF1 patients, typically attributable to megalencephaly related to increases in white-matter volume. Approximately 60% to 78% of patients with NF1 have increased signal lesions within the basal ganglia, thalamus, brainstem, and cerebellum on T2-weighted MRIs (Fig. 65.12). These areas are not routinely visible with CT. The origin and significance of these radiographic lesions are unclear, and they are referred to at times as unidentified bright objects (UBOs). Whether these MRI lesions correlate with the likelihood of cognitive impairment still is debatable; radiographic findings do not correlate with neurological deficits. Patients with NF1 tend to have full-scale intelligence quotient (IQ) scores within the low-normal range and to exhibit behavioral problems. These symptoms may be related to vascular changes in myelin sheath. Deep gray-matter radiological findings tend to decrease with time, while cortical and subcortical findings do not decrease or increase.
Clinical Features of Neurofibromatosis Type 2
Most patients who meet established diagnostic criteria for NF2 (see Table 65.3) eventually develop bilateral vestibular schwannomas, previously termed acoustic neuromas (Fig. 65.13). Symptoms of NF2 typically develop in adolescence or early adulthood but can begin in childhood. Common complaints with large acoustic tumors include hearing loss, tinnitus, vertigo, facial weakness, poor balance, and headache. Unilateral hearing loss is relatively common in the early stages. Consider screening with annual auditory brainstem responses or brain MRI.
Merlin is a novel regulator of TSC/mTORC1 signaling, such that rapamycin is being evaluated in the management of NF2 tumors (James et al., 2009).
Sturge-Weber Syndrome
The characteristic features of Sturge-Weber syndrome (SWS) are a facial cutaneous angioma (port-wine nevus) and an associated ipsilateral leptomeningeal and brain angioma. In addition to the facial nevus, other findings include mental retardation, seizures, contralateral hemiparesis and hemiatrophy, and homonymous hemianopia (Bodensteiner and Roach, 2010). However, the clinical features are variable, and individuals with cutaneous lesions and seizures but with normal intelligence and no focal neurological deficits are common. The syndrome occurs sporadically and in all races.
Cutaneous Features
The nevus typically involves the forehead and upper eyelid but also may involve both sides of the face and extend onto the trunk and limbs (Fig. 65.14). Nevi that involve only the trunk, or facial nevi that spare the upper face, rarely are associated with intracranial angioma. The facial angioma is usually obvious at birth; it may thicken over time and develop a nodular texture. Reactive hypertrophy of adjacent bone and connective tissue may occur. Some children have the characteristic neurological and radiographic features of SWS yet have no skin lesions. More frequently, the typical cutaneous and ophthalmic findings are present without clinical or radiographic evidence of an intracranial lesion. Only 10% to 20% of children with a port-wine nevus of the forehead have a leptomeningeal angioma. Although the leptomeningeal angioma is typically ipsilateral to a unilateral facial nevus, bilateral brain lesions occur in at least 15% of patients, including some with a unilateral cutaneous nevus.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


