Neurodegenerative Diseases
Juan C. Troncoso
Olga Pletnikova
In this chapter, we provide a practical approach for the assessment of neurodegenerative disease in a forensic institution. The forensic neuropathologist is frequently asked to evaluate the brains of older individuals for neurodegenerative disorders for various reasons. For example, Alzheimer disease (AD) dementia may explain why an older person wandered away from home in the middle of winter and died from exposure, and Parkinson disease (PD) can explain the history and evidence of repeated falls in other cases. In the last 20 years, as genetic factors have been uncovered in many neurodegenerative disorders, relatives have become more interested in the diagnosis of specific neurodegenerative disease because of potential genetic implications. An important caveat is that the neuropathologist can describe the presence, severity, and distribution of specific brain lesions (e.g., senile plaques or neurofibrillary tangles), but cannot say whether or not the decedent was demented. To establish the diagnosis of AD, both the pathologic changes and a clinical history of dementia are necessary.
Dementia is the most common manifestation of neurodegenerative diseases. The current definition of dementia requires the impairment of at least two cognitive domains, 1 for example, loss of memory and language deficit, such as word finding, or loss of memory and spatial disorientation. The Mini-Mental State Examination2 is a 30-point scale test commonly used to screen for cognitive deficits. Scores less than 27 points are considered abnormal. The Clinical Dementia Rating3, 4 is a functional assessment of a subject’s cognitive performance; a rating of 0 indicates normal status, 0.5 indicates suspect dementia, and 1 to 4 indicate dementia of increasing severity.
The most common causes of dementia in older adults are AD, frontotemporal dementias, Lewy body disease (including PD and dementia with Lewy bodies), and vascular disease (multiple cerebral infarcts). Not surprisingly, because these are all age-associated disorders, we often encounter them in combination. For example, it is common to find the lesions of AD and Lewy bodies in the same brain. Furthermore, there is growing evidence that the pathogenic mechanisms of these disorders may be synergistic.
ALZHEIMER DISEASE
AD is a slowly progressive disorder characterized in its early stages by prominent loss of memory, then compounded by impairments of language, praxis, judgment, orientation, and behavior. The clinical course of the disease lasts for approximately 10 years and the majority of patients require nursing home care for the last years of life. Approximately 10% of the cases are familial, secondary to mutations in the amyloid precursor protein (APP) (chromosome 21), presenilin-1 (chromosome 14), or presenilin-2 (chromosome 1) genes. These familial cases tend to have an early onset in the third or fourth decade of life. Late-onset cases (i.e., > 65 years of age) are considered mostly sporadic. Nonetheless, in this group there is a strong association with the E4 allele of the apo-E gene.5 (See review of AD genetics.6, 7, 8)
Neuropathology
In advanced cases, gross examination of the brain shows striking atrophy, with brain weights of 1, 000 g or less, prominent atrophy of the cerebral cortex and hippocampus, and marked hydrocephalus ex vacuo. In subjects in the early stages of dementia, the brain weight can be in the normal range and the cortical atrophy mild, but hippocampal atrophy tends to be easily detectable at this time (Fig. 20.1). Microscopic changes are characterized by the deposition of cerebral amyloid or β-amyloid (Aβ) in the parenchyma (Fig. 20.2) and blood vessels of the cerebral cortex (Fig. 20.3) and the development of senile plaques (Fig. 20.2) and neurofibrillary tangles (NFTs) (Fig. 20.4). Senile plaques can be divided into diffuse (Fig. 20.5) and neuritic (Fig. 20.6) plaques. The latter are used for establishing the neuropathologic diagnosis of AD according to the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD).10 Microscopic lesions of AD are traditionally examined by staining the histologic sections with silver by the Bielschowsky method or one of its many modifications.9 With this method, it is possible to distinguish NFTs and various types of senile plaques: diffuse, cored, and neuritic (Fig. 20.6). The neuritic plaques contain dystrophic neurites filled with paired helical filaments, cytoskeletal proteins, multiple vesicular organelles, lysosomes, and amyloid precursor protein.11, 12, 13 Staining with thioflavin S also allows for the detection of neuritic senile plaques and NFTs.
The histopathologic changes of AD may also be examined by immunocytochemistry for the various protein abnormalities present in this disease. Aβ antibodies identify diffuse amyloid deposits, senile plaques (Figs. 20.5 and 20.7), and vascular amyloid (Fig. 20.3). Antibodies for phosphorylated tau identify the paired helical filaments present in NFTs (Fig. 20.8), tau or neuropil threads (Fig. 20.9), and the neuritic components of the plaques (Fig. 20.10). It is also recommended to immunostain for a-synuclein to detect Lewy bodies. The advantage of using immunocytochemistry is that it is less expensive to use than silver stains; the background of the stain is cleaner, allowing a faster and unequivocal identification of lesions; and it is easier to identify alternative or coexisting
pathologies, such as tauopathy14, 15, 16 and a-synucleopathy.17, 18 The disadvantage is that current criteria for pathologic diagnosis of AD are based on silver stains and not on immunostains.
pathologies, such as tauopathy14, 15, 16 and a-synucleopathy.17, 18 The disadvantage is that current criteria for pathologic diagnosis of AD are based on silver stains and not on immunostains.
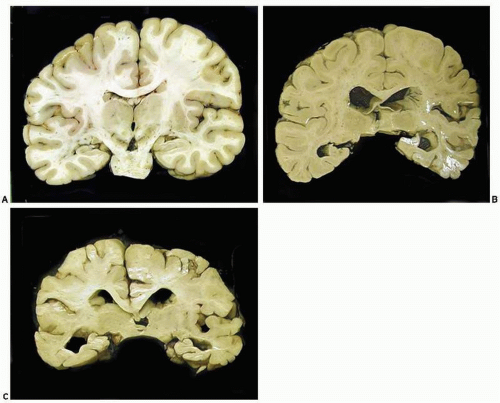 FIG. 20.1. A. Coronal sections of control brain. B. Brain of a patient with moderately advanced Alzheimer disease; note mild cortical atrophy and hydrocephalus and marked hippocampal sclerosis. C. Brain of a case of late-onset familial Alzheimer disease with severe cortical and hippocampal atrophy and marked hydrocephalus ex vacuo. |
Neuropathologic Diagnostic Criteria
Because the neuropathologic lesions of AD may also be present in the brains of cognitively normal subjects, it has been necessary to develop pathologic diagnostic criteria based on the semiquantitative assessment of neuritic plaques and NFTs. The CERAD criteria rate the density of neocortical senile plaques in a scale from none to frequent (Fig. 20.11).
The Braak classification applies to the distribution of NFTs throughout the brain, from 0 to VI.19 These pathologic descriptors, in combination with clinical information, are the bases for clinicopathologic algorithms to determine whether AD is possible, probable, or definite (CERAD), or whether the probability that the dementia can be attributed to AD is low, intermediate, or high (NIA-Reagan Institute).20
LEWY BODY DISEASES AND α-SYNUCLEINOPATHIES
These diseases are characterized biochemically by abnormalities of α-synuclein and pathologically by the presence of Lewy bodies and Lewy neurites. Lewy bodies are neuronal cytoplasmic inclusions enriched in aggregated α-synuclein and multiple other proteins. Clinically, Lewy body disease can manifest as PD or Dementia with Lewy Bodies (DLB). These two categories represent
clinicopathologic constructs, and for this reason, there is the potential for confusion in the use of the various categories and terms. To avoid this situation, we recommend establishing first the distribution and density of Lewy bodies and neurites: that is, brain stem, limbic (hippocampus, entorhinal, and cingulate cortices), or diffuse (cortical). Then it is possible to combine the pathologic findings and clinical information to arrive at a final diagnosis.
clinicopathologic constructs, and for this reason, there is the potential for confusion in the use of the various categories and terms. To avoid this situation, we recommend establishing first the distribution and density of Lewy bodies and neurites: that is, brain stem, limbic (hippocampus, entorhinal, and cingulate cortices), or diffuse (cortical). Then it is possible to combine the pathologic findings and clinical information to arrive at a final diagnosis.
 FIG. 20.2. Senile plaque with amyloid core visualized with hematoxylin-eosin stain. |
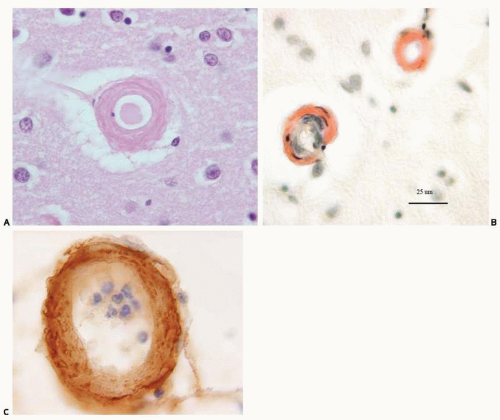 FIG. 20.3. Amyloid angiopathy. Section of the cerebral cortex immunostained with hematoxylin and eosin (A), Congo red (B), and 6E10, a monoclonal antibody for Aβ (C). Note the deposition of the amyloid within the vessel wall and also in the perivascular space. |
In this section, we will address PD and DLB, which are the most common Lewy body diseases.
Parkinson Disease
The clinical features of PD include resting tremor, akinesia, rigidity, cog-wheeling, loss of postural reflexes, poor stance and gait stability, and frequent falls. The early motor abnormalities are often unilateral. The motor abnormalities in PD are the result of the progressive degeneration of dopaminergic neurons in the pars compacta of the substantia nigra and their projection to the striatum. The disease is not limited to the striatonigral system; however, as the disease progresses, other brain regions become involved, and the majority of patients with PD develop some degree of cognitive impairment or dementia. More than 70% of patients with PD may suffer from cognitive deficits 8 years after diagnosis.21 Although for many years PD was considered predominantly a sporadic disorder, more recently, a growing number of affected kindreds have been described and causative mutations identified, such as a-synuclein, parkin, DJ-1, and LRRK2.22 As happens with other neurodegenerative disorders, familial cases of PD tend to have earlier onset than do sporadic cases. The motor manifestations of PD, tremor in particular, respond to therapy with levodopa or dopamine agonists, but these therapies do not arrest the relentless progression of the disease.
Neuropathology
The weight and external examination of the brain are usually unremarkable. Nonetheless, it is important to search for signs of trauma. On coronal sections, there is frequently depigmentation of the substantia nigra (Fig. 20.12A) and locus coeruleus. These changes may be difficult to ascertain because of the wide variation in the normal pigmentation of these nuclei. There should be no atrophy of the striatum (caudate and putamen) or lacunes of the basal ganglia. Microscopically, hematoxylin and eosin (H&E) stains of the substantia nigra show marked loss of pigmented neurons (Fig. 20.12B), pigment incontinence (Fig. 20.13), Lewy bodies (Fig. 20.14), and proliferation of reactive astrocytes.23 Careful
examination may also reveal Lewy bodies of the basal forebrain, limbic system, and association cortices. The method of choice to examine for Lewy bodies is with α-synuclein immunostains, however, which also demonstrate readily the presence of aggregated α-synuclein within neurites (Lewy neurites) and in the neuronal perikaryon, either within vesicular organelles or diffusely (Figs. 20.15 and 20.16).
examination may also reveal Lewy bodies of the basal forebrain, limbic system, and association cortices. The method of choice to examine for Lewy bodies is with α-synuclein immunostains, however, which also demonstrate readily the presence of aggregated α-synuclein within neurites (Lewy neurites) and in the neuronal perikaryon, either within vesicular organelles or diffusely (Figs. 20.15 and 20.16).
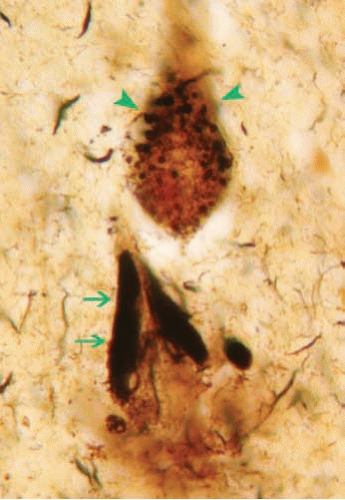 FIG. 20.4. Neurofibrillary tangles. Hirano silver stain of the hippocampus shows two pyramidal neurons bearing neurofibrillary tangles (short arrows). The neuron on the top also shows granulovacuolar degeneration (long arrow). |
α-Synuclein immunostaining has allowed mapping the whole extent of Lewy body pathology in PD, including very early lesions in the medulla24, 25 (Fig. 20.17). Many brains of patients with PD show concomitant Alzheimer lesions, in particular Aβ deposits of the cerebral cortex, and there is growing evidence to support a synergism of PD and AD.
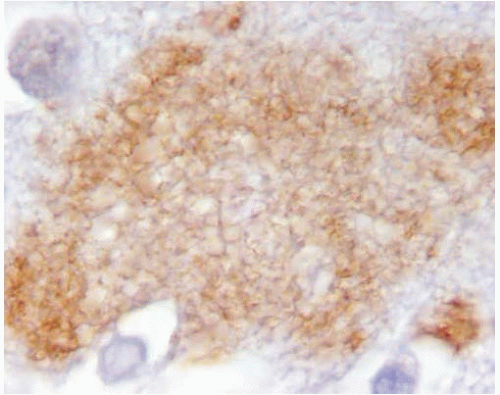 FIG. 20.5. Diffuse senile plaque recognized by immunostain with 6E10 (Signet Laboratories), a monoclonal antibody that recognizes the N-terminus of the Aβ. The diffuse plaque lacks core and dystrophic neurites. |
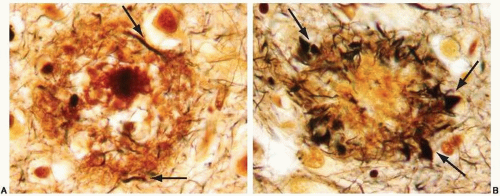 FIG. 20.6. Senile plaque with Aβ core stained with a modification of Bielschowsky silver stain method.9 A. Few dystrophic neurites are noted within the crown of the plaque (arrows); therefore, this is a neuritic plaque. B. Note the prominent and abundant dystrophic neurites in the crown of the plaques (arrows). |
Dementia with Lewy Bodies
Clinical Features
The central clinical feature of this disorder is dementia, with prominent fluctuations during the day, and visual hallucinations. The motor impairment tends to manifest after the onset of dementia and is variable in its severity, ranging from rigidity and gait instability to complete immobility. In contrast with AD, DLB tends to progress at a rather faster pace and, in our experience, patients seldom live more than 5 years after diagnosis.27
Neuropathology
Macroscopic examination of the brain is not very striking, although it may show some cortical atrophy and pallor of the
substantia nigra. Microscopically, on H&E stains, Lewy bodies are abundant in the cerebral neocortex, in particular cingulate and insular cortices, entorhinal cortex, hippocampus, basal forebrain, and substantia nigra. The pathologic changes of DLB are best appreciated on α-synuclein immunostains, however. This technique uncovers a striking amount of Lewy neurites throughout the cerebral cortex (Fig. 20.18). Currently, the pathologic diagnosis of DLB is based on consensus criterion27 that require semiquantitative assessment of Lewy bodies in brainstem, limbic regions, and neocortex. In our experience, most cases of DLB also have significant Alzheimer changes. Thus it is recommended also that silver stains or immunostains for Aβ and tau be used in the assessment of DLB.
substantia nigra. Microscopically, on H&E stains, Lewy bodies are abundant in the cerebral neocortex, in particular cingulate and insular cortices, entorhinal cortex, hippocampus, basal forebrain, and substantia nigra. The pathologic changes of DLB are best appreciated on α-synuclein immunostains, however. This technique uncovers a striking amount of Lewy neurites throughout the cerebral cortex (Fig. 20.18). Currently, the pathologic diagnosis of DLB is based on consensus criterion27 that require semiquantitative assessment of Lewy bodies in brainstem, limbic regions, and neocortex. In our experience, most cases of DLB also have significant Alzheimer changes. Thus it is recommended also that silver stains or immunostains for Aβ and tau be used in the assessment of DLB.
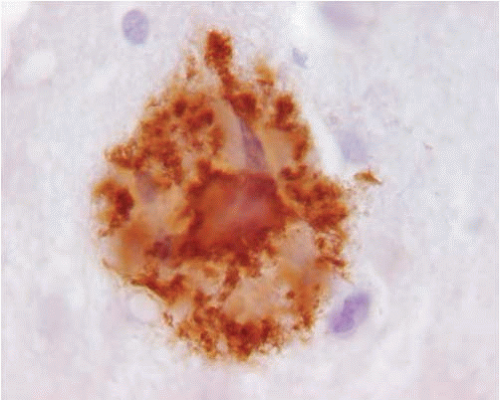 FIG. 20.7. Senile plaque immunostained with 6E10 (Signet Laboratories), a monoclonal antibody that recognizes the N-terminus of the Aβ. Note the intense staining of the core of the plaque. |
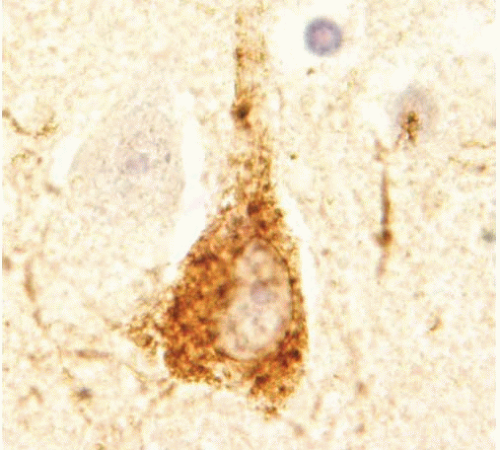 FIG. 20.8. Tau accumulation in neurons. Immunostain shows diffuse immunoreactivity in the perikaryon of pyramidal neurons. |
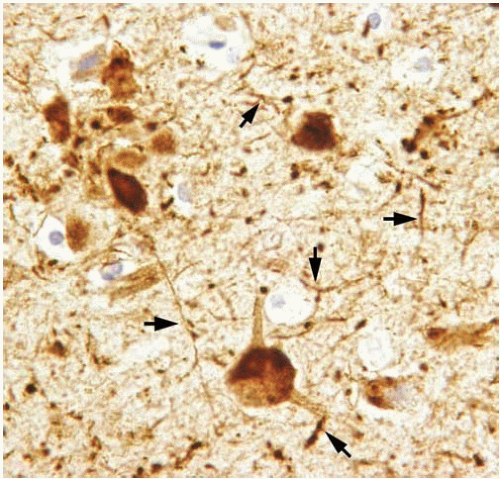 FIG. 20.9. Tau threads. Neocortex of Alzheimer disease immunostained with antibody against phosphorylated tau. Immunoreactivity is present within neuronal perikarya and also in neuritic processes. These abnormal processes are known as tau threads (arrows). |
MULTIPLE SYSTEM ATROPHY
Multiple system atrophy (MSA) is a sporadic neurodegenerative disorder characterized by α-synuclein inclusions in glial cells. It encompasses three disorders that previously were classified as independent entities: olivopontocerebellar atrophy, striatonigral degeneration, and Shy-Drager syndrome.
Clinical Features
The diagnosis of MSA is based on the presence of parkinsonism, cerebellar ataxia, autonomic failure, urinary dysfunction, and corticospinal dysfunction.28 The terms MSA-P or MSA-C are applied
to patients who have predominantly parkinsonian or cerebellar involvement, respectively.
to patients who have predominantly parkinsonian or cerebellar involvement, respectively.
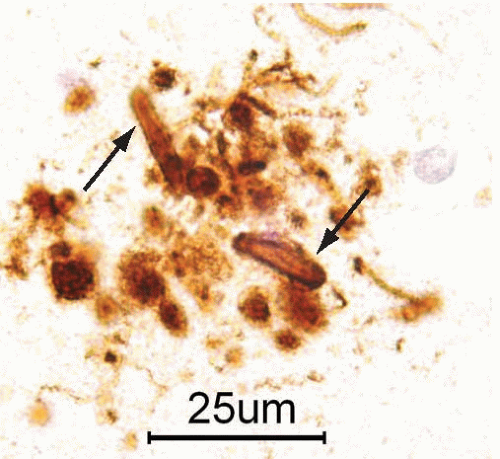 FIG. 20.10. Neuritic senile plaque immunostained with an antibody that recognizes phosphorylated tau epitopes. The immunoreactivity identifies dystrophic neurites in the crown of the plaque (arrows). |
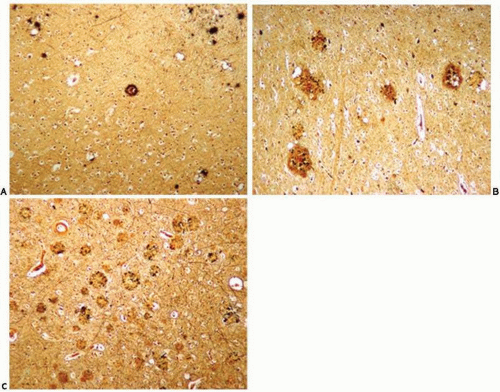 FIG. 20.11. Assessment of the frequency of neocortical neuritic plaques according to Consortium to Establish a Registry for Alzheimer Disease criteria for the pathologic diagnosis of Alzheimer Disease.10 The tissues are stained with silver method (modified Bielschowsky) and examined with a 100× magnification. A. Rare plaques. B. Moderate plaques. C. Frequent plaques. |
Neuropathology
Grossly there is no generalized atrophy, but localized atrophy is common. In MSA-P, the putamen may be atrophic and have gray discoloration, and the substantia nigra and locus coeruleus appear pale. In MSA-C, cerebellar and brainstem atrophy is common. Microscopically, there is variable neuronal loss in several regions, including putamen, globus pallidum, and substantia nigra. In the cerebellum, the Purkinje cells are more vulnerable than other neurons and their loss is most severe in the vermis. The histologic hallmark of MSA, however, is the glial cytoplasmic inclusion. This inclusion in the cytoplasm of oligodendrocytes was first described with the Gallyas silver staining method, 29 but it is more easily detected with ubiquitin or α-synuclein immunostains (Fig. 20.19). In addition to oligodendroglial inclusions, brains in MSA may also show perikaryal, neuritic, and nuclear α-synuclein inclusions in neurons.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


