Fig. 10.1
Bronchoscopic visualization of intrabronchial typical carcinoid
10.3.2.2 Histopathology and Immunohistochemistry
Typical carcinoids are characterized by organoid, trabecular, insular, palisading, ribbon, or rosette-like growth patterns separated by a fibrovascular stroma. The tumor cells are small and polyhedral with small, round, or oval nuclei and eosinophilic, finely granular cytoplasm. Mitoses are rare or absent, and the proliferative rate is usually low. Atypical carcinoids are characterized by nuclear pleomorphism, hyperchromatism, abnormal nuclear-to-cytoplasmic ratio, prominent nucleoli, and areas of increased cellularity with disorganized architecture. Mitoses are more frequent, and necroses may be found (Fig. 10.2a–c). At electron microscopy, typical carcinoids have abundant membrane-bound secretory granules, while atypical carcinoids have fewer granules, distributed in the cytoplasm [13].
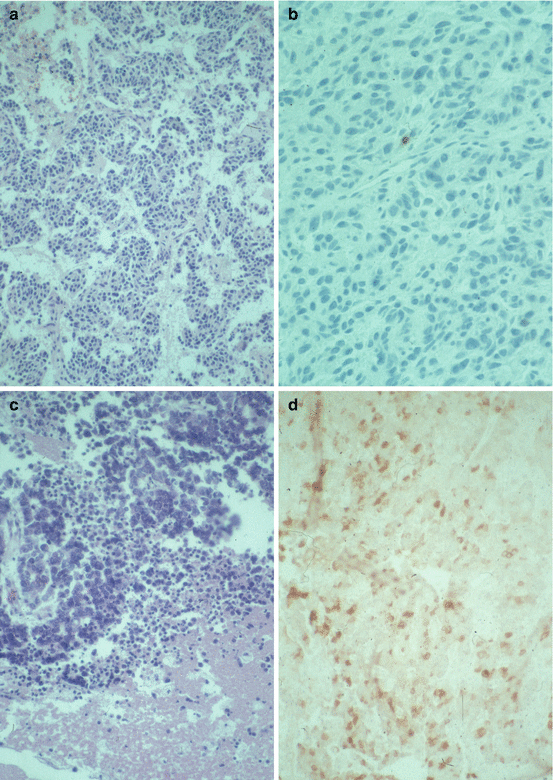
Fig. 10.2
(a) Typical lung carcinoid. Hematoxylin-eosin stain. Magnification, ×200. (b) Typical lung carcinoid. Ki67 stain, low proliferation. Magnification, ×400. (c) Atypical lung carcinoid. Hematoxylin-eosin stain. Magnification, ×200. (d) Typical lung carcinoid. Chromogranin A stain, strong positive. Magnification, ×200
Lung carcinoids are epithelial in origin and stain positive for cytokeratin. Most of the tumors also stain positive for the neuroendocrine markers chromogranin A (Fig. 10.2d), synaptophysin, and neuron-specific enolase. In addition, positive immunohistochemistry for various hormones, including serotonin, gastrin-releasing peptide (mammalian bombesin), pancreatic polypeptide, gastrin, human chorionic gonadotropin alpha subunit, leucine enkephalin, VIP, somatostatin, calcitonin, ACTH, and growth-hormone-releasing hormone is found in a majority of tumors. Positive staining for multiple hormones is not uncommon [13, 14]. Expression of S-100 protein may occur, usually in peripheral tumors [15, 16]. The standard (hematopoietic) form of the adhesion molecule CD44 (CD44s) is usually expressed in lung carcinoids and may be used for prognostic evaluation in typical carcinoids [17]. Positive staining for the retinoblastoma gene protein is also common in typical carcinoids [18, 19]. The oncoproteins p53 and bcl–2, however, are usually negative in typical carcinoids but more frequently positive in atypical carcinoids [20, 21]. Thyroid transcription factor-1 (TTF-1) is positive in 28–69 % of lung carcinoids [22, 23] and may help to differentiate a primary lung carcinoid from a metastasis from a neuroendocrine tumor originating elsewhere.
10.3.3 Genetic Alterations
Deletions in the MEN1 locus at 11q are common in both typical and atypical carcinoids [24]. Homozygous somatic inactivation of the MEN1 gene is detected in 36 % of sporadic lung carcinoids [25]. Atypical carcinoids frequently have deletions in 10q or 13q [24]. Aneuploidy has been reported in 5–32 % of typical carcinoids and 17–79 % of atypical carcinoids [4, 26–28].
10.3.4 Clinical Presentation
A substantial number of patients (13–51 %) are asymptomatic, and the tumor is diagnosed incidentally on routine chest X-ray or CT scan. Common symptoms on presentation include cough, hemoptysis, wheezing, recurrent pneumonias with or without persisting lung infiltrate on chest X-ray, dyspnea, and chest pain [14, 29–31]. Some patients may have several years’ delay in the correct diagnosis due to misdiagnosis as asthma. Like other neuroendocrine tumors, lung carcinoids may secrete hormones. Endocrine symptoms are however rare. Despite serotonin immunoreactivity is present in up to 84 % of the tumors, the classical carcinoid syndrome with flush, diarrhea, bronchoconstriction, right-sided valvular heart disease, and elevated urinary 5-hydroxyindolacetic acid is seen in only about 2–12 % of the patients, usually when liver metastases are present [32–34]. This low frequency may in part be due to the high pulmonary content of monoamine oxidase, which metabolizes serotonin, but also to the fact that most lung carcinoids do not give rise to distant metastases. An atypical carcinoid syndrome with generalized flushing, edema, lacrimation, asthma, and diarrhea may occasionally be seen. This syndrome, which must not be confused with atypical carcinoids, is caused by secretion of histamine. An ectopic Cushing’s syndrome, caused by production of adrenocorticotropic hormone (ACTH) or corticotropin-releasing factor, may be seen in 2–6 % of patients with lung carcinoids. These tumors are often small and difficult to detect with conventional radiologic imaging. Acromegaly, caused by secretion of growth-hormone-releasing hormone, is infrequent [35, 36].
Typical carcinoids metastasize in 5–20 % of patients, while up to 70 % of patients with atypical carcinoids develop metastases. Metastases most frequently occur in regional lymph nodes but also distantly to the liver, bones, brain, subcutaneous tissue, mammary glands, eyes, and adrenals [1, 32, 37, 38]. Metastases may occur late, up to 30 years after surgery for the primary tumor.
10.3.5 Diagnosis
In more than 60 % of the patients, the tumor is visible on chest X-ray. Peripheral tumors are seen as round or oval nodules. Patients with central tumors may have signs of bronchial obstruction and have peripheral atelectasis or pneumonic infiltrates which may be persistent. Computerized tomography (CT) scan (Fig. 10.3a) is more sensitive than chest X-ray and should always be performed in order to identify enlarged lymph nodes, delineate tumor growth, and detect satellite lesions (tumorlets). Magnetic resonance imaging (MRI) may be an alternative to CT scan but is less sensitive in detecting small intrapulmonary lesions.
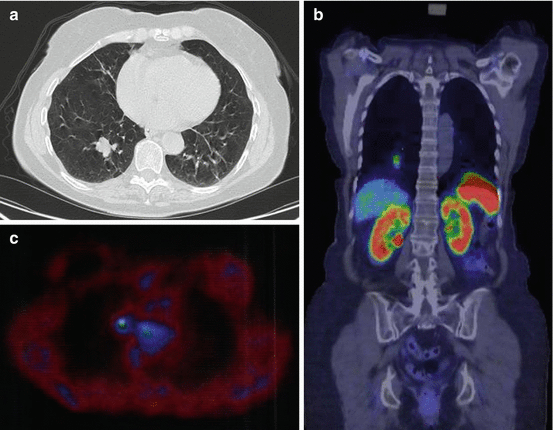
Fig. 10.3
(a) CT scan showing a carcinoid in the right lung. (b) PET with 68Ga-DOTATATE showing a carcinoid in the right lung. (c) PET with 11C-5-HTP showing a right hilar lymph node metastasis from a typical carcinoid in the right lung, 9 years after primary surgery
About 70 % of lung carcinoids express somatostatin receptors and are demonstrable on Octreoscan (scintigraphy with 111In-octreotide) [39]. Positron emission tomography (PET) with 68Ga-DOTATCOC or 68Ga-DOTATATE (Fig. 10.3b) is a newer, more sensitive method to detect somatostatin receptor-positive neuroendocrine tumors. 68Ga-PET or Octreoscan should always be performed preoperatively for staging of the disease and clarify whether these methods may be used for postoperative follow-up to detect recurrence or metastases. Patients with Cushing’s syndrome often have small tumors, which are difficult to detect on CT scan. Up to 12 years delay has been described in localizing the tumor [40]. In these patients, PET with 68Ga or Octreoscan is especially useful to obtain the correct diagnosis. An alternative is PET with 11C-5-HTP (Fig. 10.3c), which is a sensitive tool for detection of neuroendocrine tumors [41]. The value of PET with 18 F-fluorodeoxyglucose (FDG) is controversial. Some authors claim that it is of limited value in detecting lung carcinoids, since these tumors often display lower uptake than expected for malignant tumors [42, 43], while others have found this method useful in patients with both typical and atypical carcinoids [44].
Central tumors are accessible via bronchoscopy (Fig. 10.1), which is performed in most patients. Since the tumor is often covered by a layer of normal mucosa, brushing or sputum cytology is frequently negative. To obtain a preoperative diagnosis, it is thus important to take biopsies, despite the risk of bleeding. It is generally safe to take biopsies, both through a flexible and a rigid bronchoscope [45]. Peripheral tumors may be reached by transthoracic CT-guided core-needle biopsy, although misdiagnosis is not uncommon, since the differential diagnosis to small-cell lung carcinoma may be difficult. Staining the tissue sample for the proliferation marker Ki67 may aid in the differential diagnosis between these entities [46, 47].
Plasma chromogranin A should always be measured preoperatively but is usually normal if the tumor is confined to the lung. If chromogranin A is elevated, intense search for distant metastases is warranted. Measurement of urinary 5-hydroxyindolacetic acid, urinary cortisol, and plasma ACTH is not recommended routinely but is only indicated if the patient has endocrine symptoms.
10.3.6 Differential Diagnosis
Alternative diagnoses when a tumor is found on chest X-ray or CT scan include other benign or malignant lung neoplasms, hamartoma, and metastasis from another primary tumor. The histopathological differentiation, both via bronchoscopy and transthoracically, between atypical carcinoid and SCLC may sometimes be difficult, but is important since patients with carcinoids are often cured by surgery, which is generally not performed in patients with SCLC. High proliferative rate, Ki67 > 25 %, favors the diagnosis of SCLC, while a low Ki67 index indicates lung carcinoid [46, 47].
10.3.7 Treatment
The main treatment for patients with lung carcinoids is surgery, which offers the only chance for cure. External radiotherapy is mainly used for palliation of bone or brain metastases. Patients with metastatic disease or inoperable tumors can be treated with various chemotherapy combinations, peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTA-octreotate or 90Y-DOTA-octreotide, biotherapy (somatostatin analogues, alpha-interferon), or newer targeted therapies such as everolimus or sunitinib. Patients with liver metastases may undergo hepatic arterial embolization with particles, chemotherapeutic agents, or 90Y-labeled microspheres (SIR-Spheres® or TheraSpheres®). Symptomatic treatment for patients with carcinoid syndrome includes somatostatin analogues and alpha-interferon. Patients with metastatic tumors and Cushing’s syndrome are generally subjected to bilateral adrenalectomy; symptom relief may be obtained with ketoconazole, metyrapone, somatostatin analogues, or mitotane.
10.3.7.1 Surgery
Radical surgery should be considered in all patients with lung carcinoids confined to the thorax and is in most patients curative. The principles of surgery include complete removal of the primary tumor, preservation of as much healthy lung parenchyma as possible, and a thorough lymph node dissection, aided by frozen sections, with removal of all affected nodes. Possible surgical procedures include bronchotomy with resection of the tumor and bronchoplasty, sleeve resection, wedge or segmental resection, lobectomy, bilobectomy, and pneumonectomy. Wide resection margins are not necessary, but in patients with atypical carcinoids it is recommended to perform at least a lobectomy. Endoscopic removal of the tumor by YAG laser has previously not been recommended, since lung carcinoids often grow deeply into the surrounding tissue (the “iceberg” phenomenon), and radical removal thus is difficult to obtain by laser resection. Two recent studies however questioned this opinion and found initial bronchoscopic laser treatment to be a safe and effective therapy for patients with intrabronchial typical carcinoids. In 36–46 % of the patients, open surgery was however later required [48, 49]. One indication for endoscopic tumor removal by YAG laser is palliation of obstructive symptoms in patients with high cardiopulmonary operative risk and short life expectancy. In a limited number of patients, surgery may be considered after previous laser treatment to reduce the tumor and allow post-obstructive infiltrates to resolve. There are no studies showing the benefit of adjuvant treatment after radical surgery.
10.3.7.2 Radiotherapy
Patients with bone or brain metastases may have palliation from external radiotherapy. This may also be considered in cases with inoperable tumors or incomplete resection. PRRT is an interesting option in patients with metastatic or inoperable tumors and high expression of somatostatin receptors. Two uncontrolled studies have shown promising results. In a small study, 5/9 patients treated with 177Lu-DOTA-octreotate had a radiological response lasting a median of 31 months [50], and in another report, 24/84 patients (29 %) receiving 90Y-DOTA-octreotide responded radiologically [51]. Since there was a high frequency (9.5 % of all patients) of severe permanent decrease in renal function in patients treated with 90Y-DOTA-octreotide, it may be preferable to choose 177Lu-DOTA-octreotate for PRRT.
10.3.7.3 Chemotherapy
The treatment of patients with metastatic lung carcinoids depends on the histopathology (typical versus atypical carcinoid), proliferative rate (Ki67 index), somatostatin receptor expression, extent of disease, performance status of the patient, and renal and bone marrow function. Various chemotherapy combinations have been tried with limited success, including cisplatin or carboplatin + etoposide, docetaxel or paclitaxel ± doxorubicin, streptozotocin + 5-fluorouracil or doxorubicin, oxaliplatin + capecitabine, 5-fluorouracil + dacarbazine + epirubicin, and 5-fluorouracil + cisplatin + streptozotocin [52–56]. A recent study however confirmed earlier observations that temozolomide as monotherapy may be active in these patients. Significant tumor reduction was seen in 14 % and stabilization of progressive disease in 52 %. All patients with partial response had atypical carcinoids, but stabilization was observed in typical as well as atypical carcinoids [57, 58]. Combining temozolomide with capecitabine and/or bevacizumab may have better effect, but this has not been studied. These combinations are mainly used in patients with higher proliferation or if progression occurs on monotherapy with temozolomide. Patients with high Ki67 often receive platinum-based chemotherapy as first-line treatment.
10.3.7.4 Biotherapy
Biotherapy with somatostatin analogues (octreotide, lanreotide) and alpha-interferon is less effective in patients with lung carcinoids than small-bowel neuroendocrine tumors but may in some patients with low proliferation lead to control of the disease. Another indication for somatostatin analogues is symtomatic relief of a classical carcinoid syndrome or an atypical carcinoid syndrome. Sometimes, the addition of alpha-interferon is necessary to control the carcinoid syndrome.
10.3.7.5 Targeted Therapies
During recent years, new drugs targeting signal pathways or membrane receptors have proven to be active in patients with neuroendocrine tumors. Both everolimus, an mTOR inhibitor, and sunitinib, inhibiting vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), and c-kit, have shown to prolong progression-free survival in patients with pancreatic endocrine tumors. In a subanalysis from the randomized, placebo-controlled RADIANT-2 study, everolimus + octreotide LAR was found to prolong progression-free survival in patients with lung carcinoids from 5.6 months to 13.6 months compared with placebo + octreotide LAR. The difference was however not significant, p = 0.228. Minor response was seen in 67 % in the everolimus group and 27 % in the placebo group [59]. An ongoing study, which has recently completed the inclusion, is comparing everolimus, pasireotide (another somatostatin analogue), and everolimus + pasireotide in patients with well-differentiated lung carcinoids. The results are awaited with interest. Sunitinib has not been studied in lung carcinoids, but many lung carcinoids express the tyrosine kinase receptors PDGFRα, PDGFRβ, c-kit, and EGFR [60]. This makes it possible that sunitinib and other tyrosine kinase inhibitors may have activity in lung carcinoids.
10.3.8 Prognosis
Most patients with typical lung carcinoids are cured by surgery, even in the presence of lymph node metastases, and have an excellent prognosis. Five- and 10-year survivals are 87–100 % and 82–95 %, respectively [7, 14, 61–63]. Poor prognostic factors include lymph node metastases at diagnosis, presence of tumorlets, and high Ki67 index. On the other hand, positive immunohistochemistry for the standard form of the adhesion molecule CD44 (Fig. 10.4a) and positive nuclear staining for the metastasis suppressor gene nm23 (Fig. 10.4b) correlate with decreased risk for distant metastases and death [17, 64]. The prognosis for patients with atypical carcinoids is worse, although many of these patients are also cured by surgery. Five- and 10-year survivals are 40–69 % and 31–59 %, respectively [14, 30, 62, 65]. Since metastases may develop late, up to 30 years after primary surgery, patients with lung carcinoids must be followed for long time, at least 10–15 years. The follow-up should include measurement of plasma chromogranin A, CT scan of the thorax and abdomen, and PET with 68Ga-DOTATAOC/DOTATATE (in patients with somatostatin receptor-positive tumors) or MRI of the vertebral column. In selected patients, repeated bronchoscopies are indicated. Special attention should be paid to patients with atypical carcinoids or high proliferative rate.
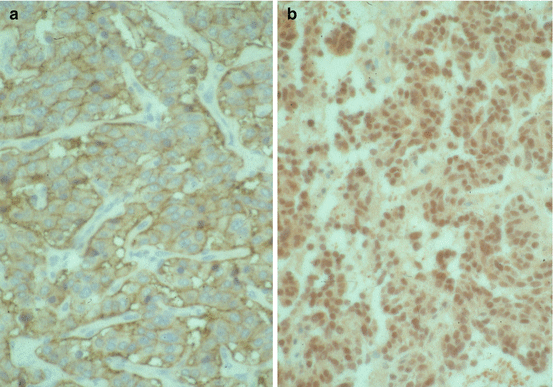
Fig. 10.4
(a) Typical lung carcinoid. CD44s stain, strong positive. Magnification, ×400. (b) Typical lung carcinoid. nm23 stain, strong positive. Magnification, ×400
10.4 Large-Cell Neuroendocrine Carcinoma
Large-cell neuroendocrine carcinomas constitute about 3 % of all lung cancers. Almost all patients are smokers. The disease is most frequent in older patients; mean age is between 59 and 66 years. Men are affected more often than women and constitute 55–86 % of the cases [4, 66, 67].
10.4.1 Pathology
LCNEC are usually peripheral and seen as large masses on X-ray. The tumor consists of large cells with moderate to abundant finely granular, eosinophilic cytoplasm and frequent nucleoli. The cells are arranged in organoid, palisading, trabecular, or rosette-like patterns (Fig. 10.5a). The mitotic rate is high, ≥11 (average 75) per 2 mm2. Large areas of necroses are frequent. The proliferative rate is high. Aneuploidy is found in 75 % of the tumors. Positive immunohistochemistry for neuroendocrine markers such as chromogranin A (Fig. 10.5b), synaptophysin, and neuron-specific enolase is less frequent than in lung carcinoids. One positive marker is enough for the diagnosis. TTF-1 is positive in about 50 % of the tumors [8]. Positive staining for p53 and bcl–2 is common [68], but retinoblastoma protein is usually not present by immunohistochemistry [19]. K–ras mutations occur in the same frequency as in other non-small-cell lung cancer [8]. At electron microscopy, the ultrastructural appearance is variable. The majority of tumors have only few cytoplasmic neurosecretory granules.
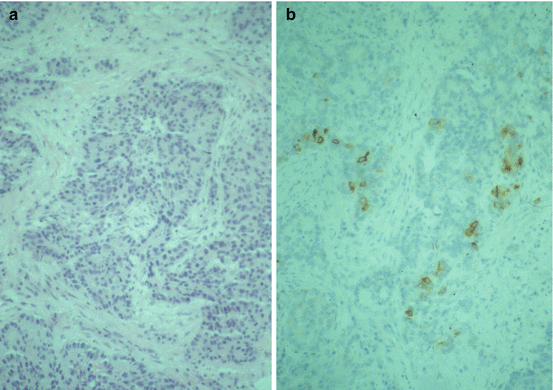
Fig. 10.5
(a) Large-cell neuroendocrine carcinoma. Hematoxylin-eosin stain. Magnification, ×200. (b) Large-cell neuroendocrine carcinoma. Chromogranin A stain, weak positive. Magnification, ×200
10.4.2 Clinical Presentation
Common presenting symptoms in patients with LCNEC include cough, dyspnea, hemoptysis, obstructive pneumonias, or constitutional symptoms such as weight loss and malaise. Some patients are asymptomatic and diagnosed incidentally by an abnormal chest X-ray. Endocrine symptoms due to ectopic hormone production have not been described. Metastases occur in between 71 and 100 % of the patients with LCNEC, most often to the hilar or mediastinal lymph nodes. Distant metastases occur to the pleura, liver, brain, bones, adrenal glands, pericardium, and abdominal lymph nodes.
10.4.3 Diagnosis
Many tumors are detectable on chest X-ray. CT scan of the chest and abdomen, as well as PET with FDG, is of value for staging. Histopathological diagnosis may be obtained by surgery or transthoracic core-needle biopsy.
10.4.4 Treatment
There is no consensus regarding the treatment of patients with LCNEC. Surgery is indicated in patients with stage 1. Since the risk for recurrence and distant metastases is high, surgery must be combined with adjuvant chemotherapy. Active combinations include cisplatin + etoposide, cisplatin + irinotecan, and nadaplatin + irinotecan. Several studies have reported the response to cisplatin-based chemotherapy to be comparable in patients with advanced LCNEC and SCLC [69–73]. Amrubicin as monotherapy has also some activity in LCNEC [74]. Patients with widespread disease may receive the same chemotherapy regimens. The role of radiotherapy is more controversial. Patients with EGFR mutations may obtain partial responses with gefitinib [75, 76]. Octreotide, which prolongs progression-free survival in patients with slow-growing neuroendocrine tumors, may be used as adjuvant treatment after surgery for LCNEC [77]. Sunitinib and other agents targeting VEGFR and c-kit are possible new drugs for treatment of LCNEC [78].
10.5 Small-Cell Lung Carcinoma
10.5.1 Epidemiology and Etiology
Small-cell lung carcinoma, which is the most aggressive form of lung malignancy, accounts for 14–20 % of all cases of lung cancer. It is most common in the seventh and eighth decades. There is a clear male preponderance, but this discrepancy may decrease due to the increasing smoking habits among women. Almost all patients with SCLC are smokers, which represents the main risk factor. Other known etiologic factors include ionizing radiation, asbestos, aromatic hydrocarbons (in particular chloromethyl ethers), and various metals [88].
10.5.2 Pathology
A majority of SCLC are centrally located, appearing as hilar or perihilar masses, spreading early to hilar and mediastinal lymph nodes [8]. Only about 5 % are seen as a peripheral solitary mass [89]. Distant metastases occur to the brain, liver, bone marrow, bones, adrenal glands, and pancreas. Brain metastases are seen in 10–20 % on presentation but occur later as the disease progresses in 25 % of patients not receiving prophylactic cranial irradiation [90]. In total, 50–80 % of the patients will develop brain metastases [91].
10.5.2.1 Light Microscopy
Small-cell lung carcinoma consists of small cells, usually less than the diameter of 3 small resting lymphocytes. The cells are round to fusiform and have a high nuclear-to-cytoplasmic ratio and finely granular, hyperchromatic nuclei with inconspicuous or absent nucleoli. Histologic patterns include trabeculae, spindling, nesting, palisading, rosettes, or solid-sheet-like growth, with indistinct cell borders. The mitotic rate is high, ≥11 per 2 mm2, median 80 per 2 mm2. Necrosis and crush artifacts are frequent. The proliferative rate is high (Fig. 10.6). Combined small-cell lung carcinoma is defined as small-cell carcinoma combined with an additional component, usually adenocarcinoma, squamous cell carcinoma, or large-cell carcinoma [8].
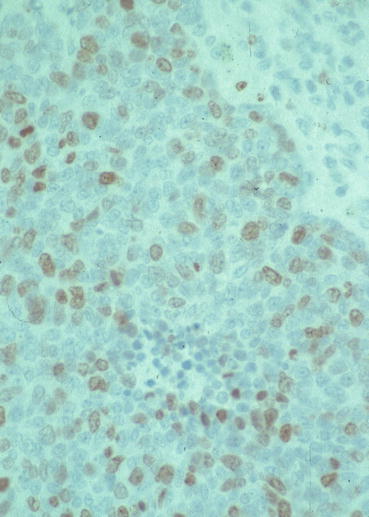
Fig. 10.6
Small-cell lung carcinoma. Ki67 stain, high proliferation. Magnification, ×400
10.5.2.2 Immunohistochemistry
The neuroendocrine markers chromogranin A, synaptophysin, and NSE stain positive in most SCLC, and TTF-1 is positive in up to 90 % of the tumors [92, 93]. Hormonal immunoreactivity, most often for ACTH, bombesin, and serotonin, may also be found, but staining for multiple hormones is less common than in lung carcinoids [4, 5]. Gastrin-releasing peptide has been shown to function as a growth factor for SCLC [94]. Positive immunostaining for p53 is common [18, 20, 68] as is bcl–2 immunoreactivity [68, 95, 96]. Retinoblastoma gene protein is usually negative at immunohistochemistry [18, 19, 97].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


