Syndrome
Gene
HNPGL
sPGL
PCC
Multiple PGL
Bilateral PCC
Malignancy risk
Parentof origin preference
MEN2
RET
Extremely rare
−
++
−
++
±
−
VHL
VHL
Rare
+
++
+
++
+
−
NF1
NF1
−
−
+
−
−
+
−
PGL1
SDHD
+++
++
+
+++
+
+
Paternal
PGL2
SDHAF2
+++
−
++
−
Not known
Paternal
PGL3
SDHC
++
+
±
+
−
±
−
PGL4
SDHB
++
+++
++
++
+
++
−
PGL5
SDHA
+
+
−
−
−
Not known
−
−
TMEM127
±
±
+++
±
++
+
−
−
MAX
−
−
++
−
++
+
Paternal
24.5 Etiology
Transcriptomic studies have shown that sporadic as well as hereditary PGLs can be divided in two main clusters linked to two different signaling pathways [53]. Cluster 1 contains all VHL (cluster 1A)- and SDHx (cluster 1B)-related tumors and is characterized by activation of the hypoxia-angiogenesis pathway in normoxia [54]. Consistent with Knudson’s two-hit hypothesis for tumorigenesis involving a tumor suppressor gene, a heterozygous germ-line mutation in an SDHx gene is usually associated with somatic loss of the nonmutant allele in the tumor, i.e., loss of heterozygosity, resulting in inactivation of SDH enzymatic activity. Inactivation of SDH has been shown to result in accumulation of succinate which acts as an inhibitor of prolyl hydroxylase (PHD) enzymatic activity. PHDs are enzymes that are required for the degradation of hypoxia-induced factor (HIF). As a consequence, even in the presence of oxygen, HIF cannot be destroyed via proteasome-mediated degradation driven by VHL protein and is stabilized to induce angiogenesis and tumorigenesis [55–57]. The latter also happens in VHL mutations. Cluster 2 contains all RET-, NF1-, TMEM127-, and MAX-mutated tumors and is associated with abnormal activation of kinase-signaling pathways, such as RAS/RAF/MAPK and PI3K/AKT/mTOR, leading to abnormal cell growth and diminished apoptosis capacity [39, 40, 58–61]. Another theory interlinking the pathogenesis of different hereditary forms of PGL is based on data from Lee et al. and Schlisio et al., showing escape from apoptosis of sympathoadrenal progenitor cells at the end of the embryonic period in case of mutations in RET, NF1, VHL, and SDH, thereby bearing the potential to form PCC/PGL in later life [62, 63]. Abovementioned data have increased overall knowledge on molecular defects in PGLs and could be used for development of new effective molecular-targeted therapies.
24.6 Diagnosis
24.6.1 Biochemistry
Synthesis of catecholamines is primarily regulated by the cytoplasmic enzyme tyrosine hydroxylase (TH), which converts tyrosine to 3,4-dihydroxyphenylalanine (DOPA) (Fig. 24.1) [64]. Tissue expression of this enzyme is largely confined to dopaminergic and noradrenergic neurons of the central nervous system, sympathetic nerves, and chromaffin cells of the adrenal medulla and extramedullary paraganglia. The following step is the production of dopamine (DA) by decarboxylation of DOPA by l-amino acid decarboxylase. DA formed in cytoplasm is transported into vesicular storage granules and is converted to norepinephrine (NE) by dopamine β-hydroxylase (DBH). In adrenal medullary chromaffin cells, NE is metabolized to epinephrine (E) by the enzyme phenylethanolamine-N-methyltransferase (PNMT). E is then translocated into chromaffin granules, where the amine is stored awaiting release. Expression of PNMT in extra-adrenal PGLs is negligent, which explains the preferential production of NE by these tumors, compared to the production of both NE and E by PCCs [65].
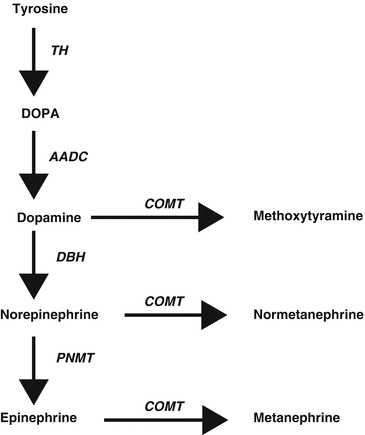
Fig. 24.1
Pathways of catecholamine synthesis and O-methylation. TH tyrosine hydroxylase, DOPA 3,4-dihydroxyphenylalanine, AADC aromatic amino acid decarboxylase, COMT catechol-O-methyltransferase, DBH dopamine β-hydroxylase, PNMT phenylethanolamine-N-methyltransferase
Catechol-O-methyltransferase (COMT) is a major player in catecholamine metabolism, catalyzing O-methylation of E to metanephrine, NE to normetanephrine, and DA to methoxytyramine.
The diagnosis of PCC/sPGL depends on the demonstration of excessive amounts of catecholamines (E, NE, DA) or their O-methylated metabolites metanephrine, normetanephrine, and methoxytyramine [66]. Since metanephrines have a longer half life and are produced continuously within tumor cells, whereas catecholamines are converted to metanephrines by the high methyltransferase activity of chromaffin tissue, measurement of plasma-free metanephrines and/or 24 h urinary excretion of fractionated metanephrines provides the best test with an excellent sensitivity of >96 % for detecting PCC/sPGL [67]. Since dietary constituents (especially caffeine and nicotine) or medication (especially acetaminophen, antihypertensive, and tricyclic antidepressant medication) can interfere with the analysis of or impact on levels of catecholamines or its metabolites, urine and plasma samples can best be collected while abstaining from these substances and/or timely stopping or changing medication (calcium entry blockers are relatively safe), with plasma being sampled in the supine position, to minimize sympathoadrenal activation [23, 68, 69]. PGLs exhibit different biochemical properties as PCCs mainly produce E and NE, sPGLs NE, and malignant PCCs NE or DA due to a dedifferentiation of the enzymatic machinery [70, 71]. Chromogranin A (CgA) is secreted from neurosecretory vesicles, along with catecholamines [72]. Plasma CgA level is often elevated in functioning and nonfunctioning PGLs [73]. The levels correlate with tumor mass, making CgA a useful tumor marker [74]. Sensitivity for identifying PGLs is 83–89 %, false-positive results however occur often due to liver or kidney failure or use of proton pump inhibitors [75]. Plasma CgA levels are particularly elevated in patients with malignant PCC [76].
24.6.2 Anatomical and Functional Imaging
Once a biochemical diagnosis of sPGL has been established, the source of catecholamine excess is subsequently localized by anatomical and functional imaging studies. Anatomical imaging such as computed tomography (CT) or magnetic resonance imaging (MRI) has an excellent sensitivity (77–98 % and 90–100 %, resp.) but lacks specificity (29–92 % and 50–100 %, resp.) for detecting PCC/sPGL [77, 78]. On CT, PCCs usually present as homogenous tumors with soft tissue density of more than 10 Hounsfield units (HU) and uniform enhancement with contrast; however, larger PCCs may undergo hemorrhage and necrosis resulting in areas of low density [79]. On MRI, PCCs present as a mass absent of fat on chemical shift, with a high signal on T2 sequences as a result of their hypervascularity [80]. MR imaging with pre- and post-contrast-enhanced 3D time-of-flight (TOF) MR angiography sequence represents the most important imaging technique for evaluation and characterization of HNPGLs [81]. MR imaging of HNPGL provides more diagnostic information than does CT scanning, because of the better soft tissue contrasts as compared to CT [82]. MRI enables multiplanar imaging of tumor extension and vessel encasement. Ultrasound has a limited diagnostic yield but can be of use in imaging HNPGL [83].
Tumors detected by anatomical imaging can subsequently be identified as PGL by functional imaging agents that specifically target the catecholamine synthesis, storage, and secretion pathway of chromaffin cells. 123I- or 131I-metaiodobenzylguanidine (MIBG) scintigraphy is the most widely used nuclear technique in first-line functional imaging of PGL (Fig. 24.2). MIBG has chemical similarities to NE and is taken up by the human NE transporter (hNET), which is expressed in most chromaffin cells and is normally responsible for catecholamine uptake [84]. The sensitivity (83–100 % for 123I – and 77–90 % for 131I-MIBG, resp.) and specificity (95–100 % for both 123I – and 131I-MIBG) are high for primary tumors; however, sensitivity of MIBG-scintigraphy for metastasis is relatively poor [77, 85, 86]. In patients with negative MIBG-scintigraphy, other tracers may be used.
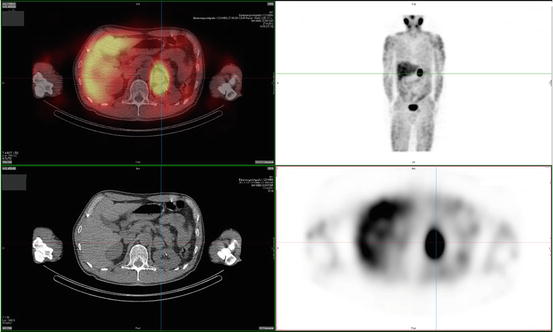
Fig. 24.2
123I-MIBG scan of a patient with a left-adrenal pheochromocytoma
PGLs have been found to express somatostatin receptor type 2, 3, and 5 on their cell surface. 111In-pentetreotide scintigraphy is of limited value in detecting PCC but may be useful in detecting sPGL and MIBG-negative metastases (Fig. 24.3) [77]. In addition, it was shown to have an excellent sensitivity of 97 % and a specificity of 83 % to detect HNPGL [87–89]. Radiolabeled dopamine (DA) or dihydroxyphenylalanine (DOPA) may be used as tracers in positron emission tomography (PET) imaging. 18 F-DOPA PET has been confirmed to be useful in the evaluation of sPGL and HNPGL (Fig. 24.4) [90]. For patients suspected for metastatic PGL, 18F-FDG or 18F-FDA PET is recommended in SDHB mutation carriers and 18F-DOPA or 18F-FDA PET in non-SDHB mutation carriers [91, 92].
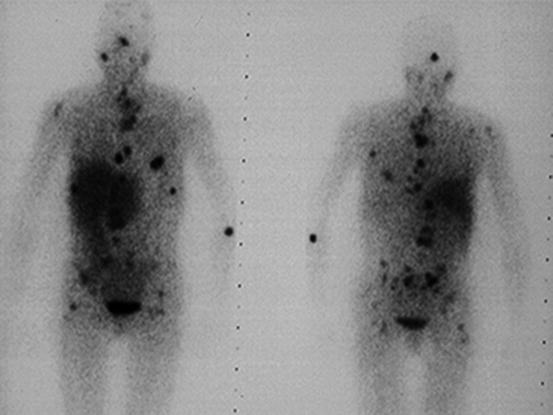
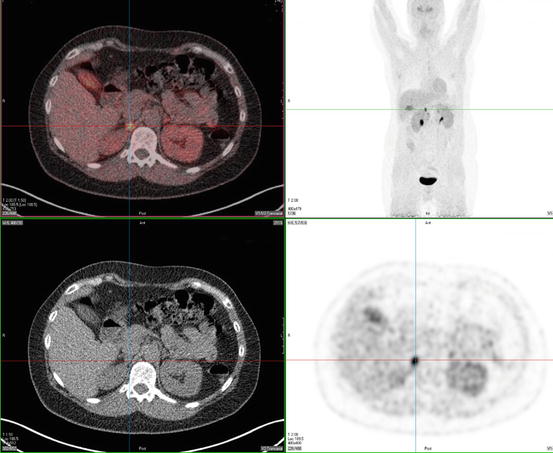
Fig. 24.3
111In-Octreoscan of a patient with diffuse metastatic sPGL of the bladder
Fig. 24.4
18F-DOPA PET scan of a patient with a right-adrenal pheochromocytoma
24.7 Treatment
The treatment of choice for PCCs and sPGLs is surgical resection, preferably laparoscopically [93], but in cases of large tumors with a high risk of malignancy, conventional laparotomy should be considered. In order to avoid surgical complications (hypertensive crisis, arrhythmias), all patients should be adequately pretreated with α-blockade (phenoxybenzamine, doxazosin), if necessary followed by β-blockade (propranolol, atenolol) and volume expansion as these patients are in a constant status of volume depletion [27]. This has been shown to reduce operative mortality to below 1 % [94]. In patients with bilateral PCC, laparoscopic cortical sparing adrenalectomies must be considered to avoid chronic glucocorticoid deficiency [95]. Preoperative injection of 123I –MIBG in combination with intraoperative use of a gamma probe may localize small lesions that are difficult to find [96, 97]. After resection of 123I –MIBG positive malignant PGL, postoperative 131I –MIBG treatment for consolidation is recommended [98]. Postoperative follow-up with biannual measurement of blood pressure and annual plasma or urinary metanephrines and anatomical imaging should be performed.
Treatment for HNPGL must be considered in relation to tumor growth velocity, biological activity of the tumor, patient age and medical condition, tumor size and site, and potential for treatment-related morbidity [17]. Because most tumors grow slowly, a wait-and-scan policy is often advised [17]. However, although HNPGLs are indolent tumors, tumor growth may lead to serious morbidity and cranial nerve impairment due to their location in close proximity to important neurovascular structures. The main treatment modalities for HNPGL are surgery and radiotherapy. A multidisciplinary team approach is recommended for the choice of treatment of most HNPGL except for the very small and easy to resect tumors. With surgery, it is possible to remove the tumor without recurrence. Preoperatively, HNPGL should be tested for catecholamine excretion, and pharmacological preparation should be started in positive cases [99]. The rate of surgical complications rises with the size of the tumor [100]. The most common complications are cranial nerve damage and vascular complications [101–104]. External beam radiotherapy and radiosurgery are alternative treatment modalities for HNPGL patients, resulting in local tumor control and sometimes regression by producing fibrosis and vascular sclerosis. These may be options in patients with large growing tumors, in which resection may result in considerable morbidity, or after incomplete resection of tumor with intracranial or skull base invasion [105].
24.8 Malignant Paragangliomas
Because there is no definite histological substrate for malignant PGL [106, 107], malignancy is defined by the presence of metastases: tumor spread in sites where chromaffin tissue is normally absent [7, 8]. Nearly 10 % of PCCs and 10–20 % of sPGLs are malignant [108], whereas HNPGLs are usually benign [109]. Malignant HNPGLs are confirmed by the presence of local metastases in cervical lymph nodes or systemic metastases, usually to bones, lung, and liver [106, 107, 109]. Malignant HNPGLs occur most frequently in patients with SDHB gene mutations [110–112]. In patients with SDHD gene mutations, malignant HNPGLs are rare [113, 114]. The primary management of patients with malignant HNPGLs should be directed toward complete surgical resection of the primary tumor and regional lymph nodes. Postoperative radiation may be beneficial in slowing the progression of residual disease [109]. Malignant PCC/sPGLs are especially associated with SDHB and MAX mutations [40, 43]. Treatment for malignant PCCs/sPGLs includes surgical debulking, pharmacological control of catecholamine-induced symptoms, external radiation, and systemic antineoplastic therapy. There is no effective treatment for malignant sPGLs. Although the usefulness of debulking of tumor has not been established, it may reduce catecholamine-related problems and improve response to further treatment. Up to 60 % of malignant PCC shows a positive MIBG uptake [115]. Treatment with therapeutic doses of 131I-MIBG resulted in an objective tumor response in 30 % of patients, stabilization of disease in 57 % of patients, and progression of disease in 13 % of patients [98, 116]. A reduction in catecholamine secretion has been seen in up to 45 % of patients. In MIBG-negative cases, patients can be treated with combination chemotherapy, of which combination of cyclophosphamide, vincristine, and dacarbazine, the so-called CVD protocol, is the most effective regimen, producing partial remissions and in single cases complete remissions, however no significant change in survival [117, 118]. In the last years, treatment with radiolabeled somatostatin analogues like 90Y-DOTATOC and 177Lu-DOTATOC has found its way in the battle against malignant PCC/PGL. Forrer et al. reported 13 stable diseases, 2 mixed responses, and 6 patients which remained progressive after treatment of 28 somatostatin receptor-positive patients with metastatic PCC/PGL with DOTATOC, without severe toxicity [119]. In addition, targeted therapies have been employed in patients with malignant PCC/PGL, of which especially sunitinib was promising in small series [120]. Conventional radiotherapy may provide effective palliation in patients with painful bone metastases; arterial embolization, chemoembolization, or radiofrequency ablation may be useful in case of liver metastases [121, 122]. Catecholamine-induced signs and symptoms can be treated with catecholamine synthesis inhibitors (alpha-methyl-para-tyrosine) or α-receptor-blocking medication. Prognosis in confirmed malignant PGL is difficult to predict but is known to be poor secondary to local recurrence or widespread metastases. Five-year survival rates are only 20–50 %; the outcome is related to tumor size [123, 124].
24.9 Genetic Testing
To date, 10 susceptibility PGL genes have been identified, so that the initial 10 % of cases classified as genetically determined has increased to 30 %. The frequency is likely to increase, as there are still young patients (with a higher likelihood of a mutation) being classified as sporadic and patients from PGL families where no mutation is found in one of the 10 susceptibility genes. Genetic testing can be of great importance for patients and their relatives because it gives the opportunity for early diagnosis through periodical imaging studies in family members of the affected individual [125]. Early detection of a familial PGL allows early detection of potentially malignant PGLs, especially in SDHB and MAX mutation carriers, and early surgical treatment, reducing the complication rate of this operation [126, 127]. Genetic testing algorithms can be made to reduce time and costs. The clinical and biochemical phenotype and SDHB and SDHA immunohistochemistry of the tumor tissue [128, 129] may be valuable tools to guide the order of genetic testing [43]. Mutation-positive carriers need regular clinical follow-up [28, 113], except for the children of female mutation carriers of SDHD, SDHAF2, and MAX, because of maternal imprinting. Although there is currently no international consensus regarding follow-up programs, annual biochemical testing, e.g., plasma-free metanephrines and/or urine fractionated metanephrines, and every 1–2 years radiological imaging of neck and/or thorax/abdomen/pelvis, depending on the mutation, with additional functional imaging in selected cases can be recommended as a surveillance program [113, 130].
References
1.
De Lellis RA, Lloyd RV, Heitz PU, Eng C (2004) Pathology and genetics of tumours of endocrine organs, World Health Organization Classification of Tumours. IARC Press, Lyon
2.
Pacak K, Keiser H, Eisenhofer G (2005) Pheochromocytoma. In: DeGroot LS, Jameson JL (eds) Textbook of endocrinology, 5th edn. Elsevier Saunders Inc, Philadelphia, pp 2501–2534
3.
Kliewer KE, Wen DR, Cancilla PA, Cochran AJ (1989) Paragangliomas: assessment of prognosis by histologic, immunohistochemical, and ultrastructural techniques. Hum Pathol 20:29–39PubMed
5.
Warren WH, Lee I, Gould VE, Memoli VA, Jao W (1985) Paragangliomas of the head and neck: ultrastructural and immunohistochemical analysis. Ultrastruct Pathol 8:333–343PubMed
6.
Lack EE, Cubilla AL, Woodruff JM (1979) Paragangliomas of the head and neck region. A pathologic study of tumors from 71 patients. Hum Pathol 10:191–218PubMed
7.
Granger JK, Houn HY (1990) Head and neck paragangliomas: a clinicopathologic study with DNA flow cytometric analysis. South Med J 83:1407–1412PubMed
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


