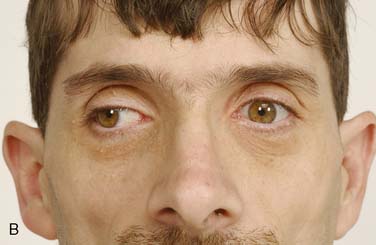
Figure 36-1 A 35-year-old man is shown. A, Note the thin body habitus and musculature build of the patient. B, Ptosis of the right eye and a left and evident internuclear ophthalmoplegia.
Laboratory evaluation revealed an elevated creatinine kinase of 2550 U/L and elevated serum pyruvate of 0.173 mmol/L (normal 0.030–0.080 mmol/L), and lactate of 4.2 mmol/L (normal 0.7–2.1 mmol/L). Magnetic resonance imaging scan of the brain demonstrated extensive diffuse T2 hyperintensity, involving the majority of the white matter, the cerebellum, and the basal ganglia bilaterally (Fig. 36-2). An electromyogram (EMG) displayed findings of a primarily demyelinating polyneuropathy with evidence of only mild axonal involvement (Tables 31-1 and 31-2). A biopsy of the left biceps showed frequent ragged red and ragged blue along with multiple cytochrome c oxidase–negative fibers, features of a mitochondrial abnormality.
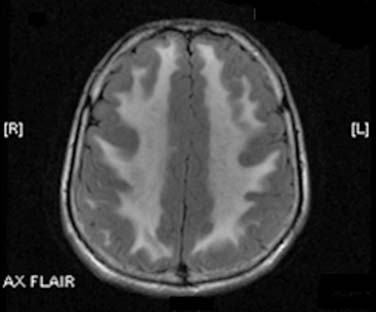
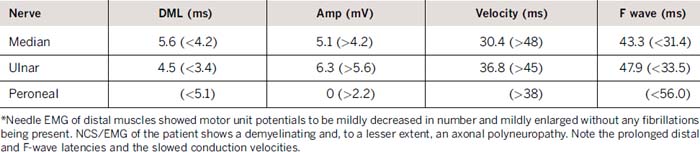
Table 36-2 Sensory Conduction Studies
| Nerve | Palmar lat (ms) | Amp (μV) |
|---|---|---|
| Median | 2.7 (<2.2) | 10.9 (>50) |
| Ulnar | 2.7 (<2.2) | 7.0 (>12) |
| Sural | 0 (<4.3) | 0 (>6) |
Given the constellation of clinical and laboratory findings involving muscle, nerve, brain, and gastrointestinal tract, a diagnosis of the mitochondrial disorder MNGIE was made. Further investigation revealed elevated levels of thymidine of 3.2 μM (normal <0.5 μM) and deoxyuridine of 8.9 μM (normal < 0.5 μM). Confirmatory genetic testing determined a homozygous T3359C mutation, shifting a leucine to proline at amino acid 285 in the gene encoding thymidine phosphorylase, the key enzyme noted to be defective in MNGIE.
CONCLUSIONS
MNGIE is a rare, multisystem autosomal recessive disorder characterized clinically by ptosis, ophthalmoparesis, peripheral neuropathy, myopathy, leukoencephalopathy, thin body habitus, and gastrointestinal dysfunction.1,2 Gastrointestinal dysmotility is a prominent manifestation, with recurrent diarrhea, borborygmi and intestinal pseudo-obstruction.1 The normal intellectual functioning of the patient suggests that the leukodystrophy involves primarily demyelination with little axonal degeneration, similar to the polyneuropathy in this disorder.1 The disease has clinically homogeneous features, although the age of onset and rate of progression varies. Patients usually die in early adulthood (mean, 37.6 years; range 26–58 years).1
The cause of MNGIE is a defect in the gene encoding thymidine phosphorylase, located at chromosome 22q13.32-qter. More than 30 mutations of this enzyme have been described.3 Thymidine phosphorylase is a cytosolic enzyme that catabolizes the nucleoside thymidine to thymine base and 2-deoxy D-ribose 1 phosphate. It also degrades deoxyuridine.3 In patients with MNGIE, plasma concentrations of both thymidine and deoxyuridine are greatly increased. Although thymidine phosphorylase is not present in mitochondria, the nucleotide pool imbalances caused by the defect likely cause mtDNA instability through an indirect mechanism.3 Despite there being both nuclear and mitochondrial genomes, replication and repair of mtDNA are solely dependent on nuclear DNA encoded genes. Mitochondria are also more dependent on the nucleoside salvage pathway compared with the nuclear genome. Therefore, it has been hypothesized that the nucleoside imbalance indirectly affects mitochondrial replication and repair, leading to the multiple deletions, depletion, and point mutations seen in the MNGIE mitochondrial genome.3
Very few studies have emphasized the clinical, neurophysiologic, and pathologic findings of the peripheral neuropathy found in MNGIE. Five patients were reported to have a clinical picture resembling chronic inflammatory demyelinating polyradiculoneuropathy (CIDP).4 In these patients, findings of fluctuating symmetric weakness progressing over greater than 2 months, hyporeflexia, elevated cerebrospinal fluid protein, and demyelination and remyelination on nerve biopsy suggested CIDP. In addition, EMG showed evidence of decreased conduction velocities, prolonged F-wave latencies, and partial conduction block. However, unlike the typical patient with CIDP, their patients with MNGIE had pathology outside of the peripheral nervous system, including gastrointestinal problems, leukoencephalopathy, and ophthalmoplegia.
Another study described several patients who had a Charcot-Marie-Tooth (CMT) presentation to their MNGIE-associated peripheral neuropathy.5 Two of the four patients described were initially misidentified as having CMT due to bilateral pes cavus and distal lower extremity weakness. EMG demonstrated a demyelinating peripheral neuropathy with axonal loss in both patients. Nerve biopsy showed reduced myelinated fiber density with regenerating fiber clusters and a few onion bulbs. Teased fiber preparations revealed evidence of demyelination and remyelination with occasional tomaculae. Other case series considered the neuropathy mild in most cases.6 Trigeminal neuralgia has also been described.7 Therefore, there is phenotypic variation in the MNGIE-associated peripheral neuropathy, ranging from a relatively mild sensory neuropathy to more severe cases resembling CMT and CIDP.
In our patient, the low nerve conduction values indicate that demyelination was a major feature of the polyneuropathy. The small or absent sensory nerve action potentials suggested the presence of mild axonal degeneration, as did the presence of a mild decrease in motor unit potentials and their mild enlargement without evidence of fibrillation potentials.
There is some promise that MNGIE may be a treatable mitochondrial disease. It has been shown that peritoneal dialysis stopped vomiting and reduced abdominal pain in one patient, although plasma nucleoside levels were not affected.8 Two other studies showed that the infusion of healthy platelets and carrier erythrocyte entrapped thymidine phosphorylase transiently restored thymidine phosphorylase, which reduced thymidine and deoxyuridine levels.9,10 Furthermore, more permanent treatments, such as allogenic stem cell transplant, have been shown to correct the biochemical derangements, although the clinical affect of such treatment is still unknown.11
In conclusion, MNGIE is a multisystem mitochondrial disorder characterized by demyelination of peripheral and central axons, gastrointestinal dysfunction, ophthalmoplegia, ptosis, and myopathy. It is an autosomal recessive disorder caused by a defect in the gene encoding thymidine phosphorylase. There is wide variability in the phenotypic presentation of the neuropathy. However, there is promise that MNGIE may soon be a treatable mitochondrial disease.
Related posts:
 Malignant Peripheral Nerve Sheath Tumor
Disseminated Sporotrichosis with Multiple Granulomatous Mononeuropathies
Late Sporadic CMT4C—A New KIAA1985 Mutation
Late-Onset Transthyretin Val30Met Familial Amyloid Polyneuropathy Unrelated to Endemic Foci
A Weak, and Numb Patient with Tremor—Antimyelin-Associated Glycoprotein Polyneuropathy
Copper Deficiency Myeloneuropathy
Malignant Peripheral Nerve Sheath Tumor
Disseminated Sporotrichosis with Multiple Granulomatous Mononeuropathies
Late Sporadic CMT4C—A New KIAA1985 Mutation
Late-Onset Transthyretin Val30Met Familial Amyloid Polyneuropathy Unrelated to Endemic Foci
A Weak, and Numb Patient with Tremor—Antimyelin-Associated Glycoprotein Polyneuropathy
Copper Deficiency Myeloneuropathy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


