Chapter 39 Neurotransmitter-Related Disorders
For ease of classification, these disorders can be divided into five groups:
Monoaminergic Neurotransmitter Deficiency States with Hyperphenylalaninemia
Overview
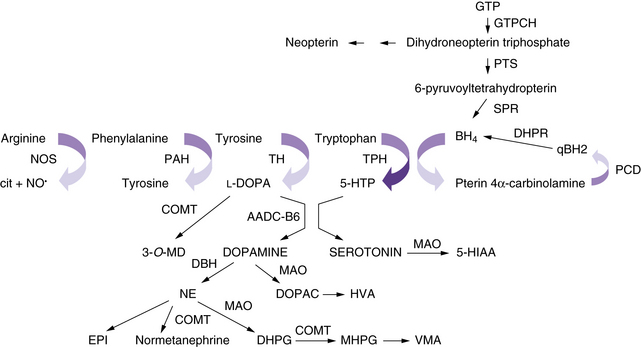
The neurotransmitter deficiency in infants in this group arises as a result of defects in BH4 metabolism (Figure 39-1). Patients are usually identified by elevated phenylalanine levels on newborn screening, as BH4 is required for phenylalanine hydroxylation in the liver. The accompanying neurotransmitter deficiency results from the lack of BH4, an obligatory co-factor required for the synthesis of catecholamines and serotonin. Although most academic biochemical genetics clinics that monitor children with phenylketonuria systematically perform the additional studies required to diagnose this group of disorders, children occasionally are not identified until they have progressive neurologic symptoms or clear evidence of developmental delay despite a phenylalanine-restricted diet. In the past, these patients were referred to as atypical phenylketonurics. In some cases, such afflicted infants are overlooked because screening is performed before an adequate interval of protein intake, resulting in a false-negative result on newborn testing.
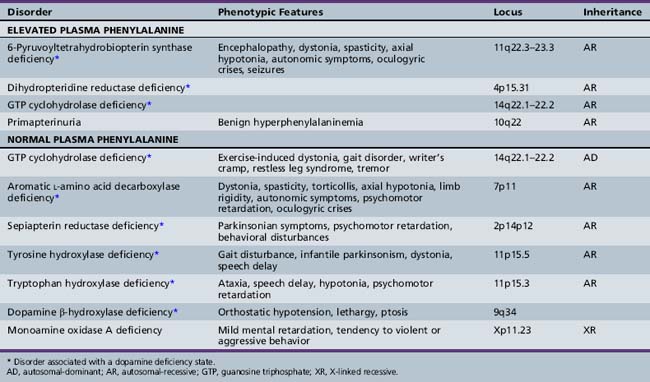
Approximately 1–3 percent of patients with hyperphenylalaninemia have an associated BH4 deficiency state. It is critical to identify such children so that BH4 and neurotransmitter precursors can be supplemented as early as possible. The two most commonly identified disorders in children presenting with hyperphenylalaninemia in the newborn period are 6-pyruvoyltetrahydropterin synthase deficiency, accounting for 60 percent of mutations in atypical phenylketonurics, and dihydropteridine reductase deficiency, affecting 30 percent of cases [Longo, 2009]. 6-Pyruvoyltetrahydropterin synthase deficiency results in inadequate BH4 synthesis; dihydropteridine reductase deficiency results in decreased regeneration of BH4 from dihydrobiopterin (see Figure 39-1). Both are autosomal-recessive disorders in which hyperphenylalaninemia results from a deficiency of BH4. Because of the involvement of BH4 in catecholamine and serotonin synthesis, such infants also have a manifest deficiency of neurotransmitter metabolites in addition to hyperphenylalaninemia. Other conditions in this category include autosomal-recessive guanosine triphosphate cyclohydrolase (GTPCH) deficiency and primapterinuria (Table 39-2).
Role of BH4 in the Central Nervous System
Because BH4 is required for the hydroxylation of aromatic amino acids, its importance in the central nervous system (CNS) becomes immediately apparent. Tyrosine and tryptophan are required for the synthesis of catecholamines and serotonin. A BH4-dependent process can be strongly suspected when plasma phenylalanine levels return to normal after BH4 supplementation. A dose of 5–10 mg/kg of BH4 is the usual recommended dose for correction of peripheral hyperphenylalaninemia, and 10–20 mg/kg per day has been recently advocated in classic phenylketonuria. Because BH4 crosses the blood–brain barrier poorly, lifelong supplementation with the neurotransmitter precursors l-DOPA and 5-hydroxytryptophan, along with carbidopa to prevent peripheral decarboxylation, is necessary in most of the disorders mentioned earlier. A much higher dose of BH4, approximately 20 mg/kg, can return BH4 levels in the cerebrospinal fluid to normal, but it remains prohibitively expensive, and no studies exist as to the possible additional benefit of such a regimen. Dosing can be monitored by assessing clinical response and periodically measuring cerebrospinal fluid levels of neurotransmitter metabolites [Hyland, 2007]. Verifying normalization of serum prolactin levels has been advocated, but sensitivity is limited, as is detection of overadministration of precursors [Concolino et al., 2008]. The greater requirement of tyrosine hydroxylase for BH4 in comparison with tryptophan hydroxylase may explain the more severe impairment in the catecholaminergic system compared with the serotonergic system.
6-Pyruvoyltetrahydropterin Synthase Deficiency
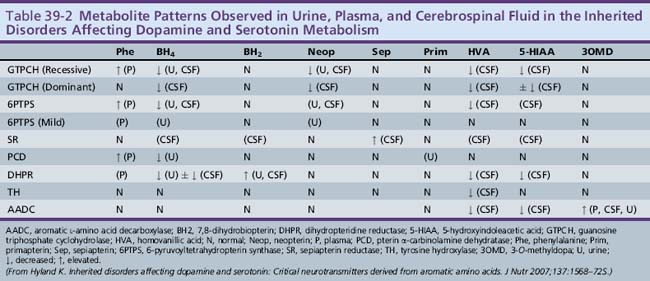
6-Pyruvoyltetrahydropterin synthase catalyzes the elimination of inorganic triphosphate from dihydroneopterin triphosphate to form 6-pyruvoyltetrahydropterin. Patients have elevated neopterin to biopterin ratios in urine and plasma. Reduced 6-pyruvoyltetrahydropterin synthase activity can be documented in red blood cells. In the classic form of the disorder, patients have reduced catecholamine and serotonin metabolites and an increased neopterin to biopterin level in cerebrospinal fluid. Patients are usually detected by newborn screening, as with phenylketonurics. They exhibit progressive signs of neurologic involvement in the first few months of life, including extrapyramidal signs, axial and truncal hypotonia, hypokinesia, feeding difficulties, choreoathetotic or dystonic limb movements, and autonomic symptoms. Many of these patients, despite early diagnosis and supplementation with BH4 and neurotransmitter precursors, continue to manifest delay in development [Dudesek et al., 2001]. A “peripheral” form of the disorder is associated with nearly one-third of known mutations with the human PTS gene [Thony and Blau, 2006], and is characterized by normal central neurotransmitter levels and less significant or transient hyperphenylalaninemia [Niederwieser et al., 1987]. Patients with the peripheral form have an excellent prognosis for normal neurologic development, provided the hyperphenylalaninemia is corrected by diet or BH4 administration.
Dihydropteridine Reductase Deficiency
Dihydropteridine reductase deficiency manifests in a variety of phenotypes, all with hyperphenylalaninemia. The clinical presentation is similar to that of central 6-pyruvoyltetrahydropterin synthase deficiency. Without folinic acid to restore methyltetrahydrofolate status in the CNS, these patients can have progressive calcification of the basal ganglia and subcortical regions, despite treatment with BH4 and neurotransmitter precursors [Woody et al., 1989]. A juvenile variant has been reported in which siblings were developmentally normal until 6 years of age, at which time they developed progressive encephalopathy, epilepsy, and pyramidal, cerebellar, and extrapyramidal features on clinical examination [Larnaout et al., 1998]. Diagnosis can be confirmed by the pattern of urine pterins and documentation of abnormal dihydropteridine reductase activity in skin fibroblasts [Milstien et al., 1980]. Results of phenylalanine loading tests are abnormal, and phenylalanine status improves or returns to normal with BH4 supplementation. Cerebrospinal fluid neurotransmitter and pterin analysis reveals reduced concentrations of homovanillic acid and 5-hydroxyindoleacetic acid, decreased or normal BH4 levels, and elevated dihydrobiopterin levels.
Pterin-4α-Carbinolamine Dehydratase Deficiency (Primapterinuria)
Pterin-4α-carbinolamine dehydratase deficiency, or primapterinuria, is a cause of mild hyperphenylalaninemia [Blau et al., 1988; Adler et al., 1992]. These infants are usually identified on newborn screening but generally have a benign course with normal development. Phenylalanine hydroxylase catalyzes the conversion of phenylalanine to tyrosine, during which BH4 is converted to the unstable carbinolamine 4α-hydroxytetrahydrobiopterin [Citron et al., 1993]. Carbinolamine dehydratase catalyzes the dehydration of carbinolamine to quinonoid dihydropterin (qBH2). The decreased rate of dehydration is responsible for the production of 7-biopterin in some mildly hyperphenylalaninemic individuals [Thony et al., 1998]. Urine studies reveal an excess of 7-substituted pterins, reduced biopterin levels, and an increased neopterin to biopterin ratio.
Monoaminergic Neurotransmitter Deficiency States without Hyperphenylalaninemia
Segawa’s Disease or Autosomal-Dominant Dopa-Responsive Dystonia
The best-described and most widely identified entity among this group of disorders is autosomal-dominant dopa-responsive dystonia caused by GTPCH deficiency, or Segawa’s disease [Segawa et al., 1976]. Identification and treatment of this disorder can be extremely rewarding because patients often benefit greatly from directed treatment of the associated dopamine deficiency state [Harwood et al., 1994]. Patients with a classic presentation of exercise-induced dystonia are not difficult to recognize. This diagnosis should also be considered in patients with spastic diplegia, especially when significant fluctuation in gait or worsening gait at the end of the day is noted, and in patients with more atypical presentations, including writer’s cramp, asymmetric limb dystonia, tremor, or restless leg-type symptoms. In patients with a classic presentation, many clinicians can make the diagnosis on a presumptive basis, after observing remission of symptoms with a trial of l-DOPA/carbidopa.
As of 2009 there are more than 85 known mutations of the GCH-1 gene, and in a large review of dopa-responsive dystonia, it was found that 60 percent are point mutations [Clot et al., 2009]. Although inheritance is autosomal-dominant, penetrance is incomplete, and variable expressivity among family members with the same mutation is well documented [Steinberger et al., 1998]. For instance, one might see spastic diplegia, writer’s cramp, restless leg syndrome, and more typical exercise-induced dystonia phenotypes among different members of the same family. The female to male ratio in sporadic cases is 4:1, and investigators have confirmed increased penetrance of GTPCH-I gene mutations in females [Ichinose et al., 1994; Furukawa et al., 1998]. Of note, a review of 47 patients with GCH-1 mutations or deletions found 40 patients manifested dystonic symptoms in the first decade of life [Clot et al., 2009].
Not surprisingly, mood and sleep disorders appear to be unusually prevalent in families with the disorder. Recent study of the neurologic and psychiatric phenotype of affected individuals within three extended families showed a higher frequency of major depressive disorder and obsessive-compulsive disorder, as well as disrupted sleep in 55 percent of patients [Van Hove et al., 2006]. These data help to support the idea that the disease carries an increased burden of attentional difficulties, anxiety, dysphoria, depression, and sleep disorders.
Aromatic l-Amino Acid Decarboxylase or Dopa-Decarboxylase Deficiency
Aromatic l-amino acid decarboxylase is a pyridoxine-dependent enzyme that decarboxylates l-DOPA and 5-hydroxytryptophan to make dopamine and serotonin, respectively. Patients with this disorder typically present in the first few months of life with dystonia or intermittent limb spasticity, axial and truncal hypotonia, oculogyric crises, autonomic symptoms, and ptosis [Hyland et al., 1992]. Neonatal symptoms, including poor suck and feeding difficulties, ptosis, lethargy, and hypothermia, are common. Neurologic signs and symptoms are clearly evident within the first few months of life in all patients described to date. These patients demonstrate multisystemic involvement with a wide array of neurologic difficulties, including problems with sleep, attention, emotional regulation, and cognitive function that extend well beyond their motor difficulties. As they get older, gross motor delays with fluctuating tone, ataxia, and expressive speech impairment are prominent features, even in the patients with the best outcomes.
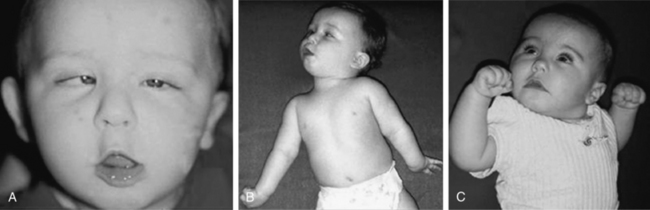
The phenomenology of the movement disorder is remarkably similar among the cases and, not surprisingly, shares a number of features in common with children with BH4 deficiency disorders, such as 6-pyruvoyltetrahydropterin synthase deficiency and dihydropteridine reductase deficiency, and the autosomal-recessive form of GTPCH deficiency. Intermittent oculogyric crises and limb dystonia, generalized athetosis, and an overall paucity of voluntary movement become evident between 1 and 6 months of age. Tongue thrusting, ocular convergence spasm (Figure 39-2), myoclonic jerks, and episodes of sudden loss of head control or episodes resembling flexor spasms are common and frequently lead to a clinical diagnosis of epilepsy. Oculogyric crises, orofacial dystonia, torticollis, limb tremor with attempted voluntary movement, and blepharospasm are often the most compelling evidence supporting a defect in dopaminergic transmission. Breath-holding or apneic spells, paroxysmal sweating, nasal congestion, sudden respiratory or cardiorespiratory arrest, unresponsiveness associated with hypoglycemia, intermittent hypothermia, and feeding and gastrointestinal issues are manifestations of the often profound autonomic dysfunction these patients demonstrate [Swoboda et al., 2003]. First- and second-degree relatives of affected individuals have been shown to have a high incidence of psychiatric disorders, including depression, psychosis, suicide, or suicide attempts [Manegold et al., 2009].
Cerebrospinal fluid neurotransmitter metabolites demonstrate a characteristic pattern: low homovanillic acid and 5-hydroxyindoleacetic acid levels; markedly elevated 3-O-methyldopa, 5-hydroxytryptophan, and l-DOPA; and normal biopterin and neopterin levels. Plasma l-DOPA is markedly elevated. Urine catecholamines may be reduced or elevated, specifically with vanillactic acid, despite normal preliminary organic acid results [Manegold et al., 2009; Abeling et al., 1998, 2000].
Although some children benefit in terms of the underlying movement disorder, treatment is complex, and these patients are vulnerable to an array of medication-related side effects. Instead of replacement of neurotransmitter precursors, as in the BH4 deficiency-related disorders, the use of neurotransmitter receptor agonists or strategies to hinder reuptake or metabolism of endogenously produced neurotransmitters is necessary. Reported benefit has been noted in a subset of patients with monoamine oxidase inhibitors, dopamine receptor agonists, anticholinergic agents, pyridoxine, and, in rare cases associated with a defect in the gene encoding aromatic l-amino acid decarboxylase at the dopa-binding site, l-DOPA [Swoboda et al., 1999]. Dopamine receptor antagonists, such as clozapine, have been proposed as a new therapeutic option, given their increase in aromatic l-amino acid decarboxylase activity in a mouse model [Allen et al., 2009].
However, in spite of a variety of treatment interventions directed at ameliorating the effects of the associated neurotransmitter deficiency state, overall clinical outcomes in aromatic l-amino acid decarboxylase deficiency remain poor. All patients have had some degree of cognitive impairment, and there is increasing evidence of a gender-dependent discrepancy in severity, with females demonstrating the most severe phenotypes [Pons et al., 2004].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


