Although generalized convulsive status epilepticus has been recognized for millennia, nonconvulsive status epilepticus (NCSE) is a much more recent concept. Perhaps by association with other forms of epilepsy with altered consciousness, some speculation about the possible existence of NCSE emerged by 1800 or so. Trousseau, at Hotel Dieu in Paris, noted that petit mal seizures might recur with sufficient frequency “that one seizure would become confused with the next, simulating a continuous seizure which might persist for two or three days millennia” thus anticipating a diagnosis of absence status epilepticus (1, 2 and 3). Nonconvulsive spells also attracted the attention of Jules Falret (1824 to 1902) who said that a patient with “petit mal intellectuel … might leave home or work, with clouded mind … [and] … complete lapses of memory (2,4).”
Samuel Wilks in London in the late 1800s described a patient who was: “… in the condition which is popularly called ‘lost;’ he is scarcely conscious of acts and conversation going on around him, yet he may continue walking in a given direction, showing that his movements must still, in a measure, be guided by his senses … in much the same state as a somnambulist. This condition … is called the status epilepticus, although the term is more usually applied to [a] patient … who, after a succession of fits, lay for hours in a state of lethargy. In the milder forms it is one of great interest from a physiology point-of-view and seems to point to the possibility of a subconscious state, in which the brain is sufficiently active to control the spinal system and yet not awake [for] consciousness (1,5).”
Also in the late 1800s, Charcot and others described cases suggestive of NCSE. Charcot postulated that a state of somnambulism derived from ongoing seizures. He presented a patient with a nonconvulsive form of long-lasting seizures, several times in his “Leçons du Mardi” at the Saltpêtrière—a delivery man who wandered about Paris and even to the coast, at Brest, being arrested on one occasion and released only on the cognizance of Charcot—who treated him with bromides (6). In Britain, Gowers speculated that similar states rather than being ictal, occurred after the seizure: “After epileptic fits of moderate severity, the patient may pass into a condition of mental automatism, in which various acts are performed in an apparently conscious manner, but of which no recollection is afterward retained (7).”
Charcot’s observations fostered speculation about the existence of NCSE, but without an appreciation of the role of electricity in the nervous system [starting to emerge in the 17th century (8)], it would be hard to conceive a modern understanding of NCSE. Clinical descriptions of confusional states (even with signs of epilepsy such as twitching of facial or limb muscles) were insufficient to prove that NCSE was a proper diagnosis. Encephalopathy, psychogenic unresponsiveness, or postictal behavior might appear very similar.
Berger’s 1929 invention and development of the electroencephalogram (EEG) made it possible to record the previously suspected electrical activity of the brain (9). He also began studies correlating EEG activity, as a measure of brain electrical function, with psychologic and behavioral abnormalities, and eventually, epileptic seizures (10). Thus, earlier historical descriptions of what might have been NCSE were thrust into a provable domain. Just over a decade later, clinical-EEG correlations showed beyond doubt that NCSE was part of epilepsy and not, as some had suspected, from hysterical or nonepileptic fugue states. The stage was set for modern electroclinical correlation, including video-EEG monitoring, various forms of imaging, and refinement of treatment.
Lennox detailed one of the first cases of “petit mal status” with an electrographic correlate in 1945 (11). In 1954, Penfield and Jasper described simple partial status epilepticus (SPSE) or aura continua in the form of recurrent sensory phenomena, commenting that they were “at least as common as continuing circumscribed movements (12).” In the first thorough description of complex partial status epilepticus (CPSE) in 1956, Gastaut and Roger described a nurse whose seizures may have lasted for months (13). In the past half century, there have been numerous clinical series detailing the electroclinical correlates and nature of NCSE.
DEFINITION
It is difficult to state a definition of NCSE, primarily because the clinical manifestations are protean and because the EEG findings, even while crucial for diagnosis, are often ambiguous or controversial. The significance of epileptiform waveforms and other abnormalities on the EEG is often unclear, and even the role of clearly epileptiform EEG abnormalities in causing clinical deficits can be uncertain or impossible to determine. Because NCSE is considered an epileptic process without convulsions (or no more than minimal motor manifestations), almost all definitions insist on EEG evidence of continuing or very frequent epileptiform activity.
The definition of generalized convulsive status epilepticus (GCSE) can be complicated and controversial (Chapter 28), but that for NCSE is much more so. It has been defined as a “range of conditions in which electrographic seizure activity is prolonged and results in nonconvulsive clinical symptoms. [It is …] primarily as a form of epileptic cerebral response which is dependant largely on the level of cerebral development and integrity, the presence or absence of encephalopathy, the type of epilepsy syndrome, and the anatomical location of the epileptic activity (14).” This definition reflects the pleomorphic character of NCSE and alludes to important insights into its biology, but diagnosis of individual cases can remain challenging. While there have been several proposals to change a temporal definition of GCSE from 30 to 5 or 10 minutes (15), it is probably most reasonable to use a 30-minute definition for NCSE (16).
CLINICAL PRESENTATION
NCSE has remarkably varied and often very subtle presentations (17,18). At times, it follows generalized seizures and GCSE, that is, GCSE continues in a more subtle form; the epileptiform discharges of a seizure continue on the EEG after the motor manifestations (convulsions) have ceased. In this case, it is best to consider NCSE as a later stage of GCSE, in terms of its pathophysiology, clinical implications, and mandate for treatment. Any patient who has not recovered from a seizure within 20 to 30 minutes or so should be considered as possibly in NCSE.
NCSE can also occur without clinically evident seizures, that is, purely nonconvulsive events. Even on its own (not following GCSE), NCSE has pleomorphic presentations. Its behavioral correlates in different types of NCSE arise from—or are at least associated with—different areas of maximal involvement of seizure activity in the brain.
NCSE typically encompasses an ictal impairment of mental status, cognition, or behavior compared with a baseline state; automatisms; or change in sensory perception (e.g., auditory, visual, somatosensory, or psychic) (19). Anterograde and retrograde memory may be affected; affects can be altered (agitation, sadness, weeping); speech and language may be impaired (with mutism or paraphasic errors); and confusional states may occur. There can be psychomotor retardation or behavioral changes, beyond the patient’s baseline. Sometimes, there are admixed, but generally minimal, motor manifestations, such as subtle facial or limb jerking, blinking, eye deviation with nystagmus (20), or perioral and eyelid myoclonus—highly suggestive of NCSE in the appropriate clinical setting (21).
Most epidemiologic studies have been focused on the incidence of GCSE. All forms of NCSE may constitute one-fourth of all cases of SE (22), and thus have an incidence of about 10/100,000 persons per year in the general population (16). About half of all cases in a German study of SE were nonconvulsive, implying about 8.5 incident cases per 100,000 population per year (23). Given the difficulties in diagnosis, these estimates of the incidence of NCSE may be far too low.
DIFFERENTIAL DIAGNOSIS
NCSE often occurs in patients with underlying chronic medical and neurologic illnesses, and also with acute and subacute medical problems such as infections, vascular disease, or metabolic abnormalities. The other illnesses may obscure the diagnosis. Many patients with NCSE have had their compromised cognition attributed to problems such as electrolyte imbalance, hyperglycemia, pneumonia, or earlier seizures (24). Many patients have dementia, mental retardation, or psychiatric disorders, making it difficult for unfamiliar observers, including physicians, to notice a new perturbation in cognition or behavior (19,24, 25, 26, 27 and 28). It is often difficult to determine if the patient is “more confused than usual,” “confused beyond what might be attributable to an infection or metabolic disturbance,” or “inappropriately lethargic after a convulsion.”
Altered alertness or behavior may be ascribed to an encephalopathy or a postictal state, delaying diagnosis (19,24). Patients with chronic psychiatric illness are at risk of delayed diagnosis of NCSE because of their neuroleptic burden and propensity for starting and stopping benzodiazepines (BDZs)—triggers for de novo NCSE (29). Especially in the elderly, NCSE may present with confusion, suggesting medication side effects or dementia (30,31). Many sick, hospitalized patients have altered mental status due to medications, infections, and metabolic encephalopathies from their underlying illnesses, and the same illness may cause or precipitate NCSE (32,33). Diagnostic clues of NCSE include an earlier history of epilepsy or risk factors for epilepsy.
NCSE is typically discovered in the emergency department, intensive care units (ICUs), and on neurology and psychiatry services, although it can occur during other hospital services and even in ambulatory patients. In the emergency department, the diagnosis of NCSE may be missed when there is lethargy or confusion attributed to a postictal state; ictal confusion mistaken for a metabolic encephalopathy; unresponsiveness and catalepsy presumed to be psychogenic; obtundation thought to be due to alcohol or drug intoxication; hallucinations and agitation mistaken for psychosis or delirium; lethargy presumed due to hyperglycemia; mutism attributed to aphasia; and laughing and crying ascribed to emotional lability (24). In hospitalized or institutionalized patients, NCSE can present primarily as an impaired level of consciousness or responsiveness.
Drugs precipitating some cases of NCSE include neuroleptics, cephalosporins, radiologic contrast agents (34), and gamma amino butyric acid (GABA) agonists, including baclofen (35), tiagabine, and vigabatrin (36,37). Occasionally, refractory NCSE can be caused by mitochondrial disorders, including mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) (38). Unusual forms of NCSE may result from paraneoplastic syndromes, especially in those likely caused by cell surface antibodies (39). In the anti-NMDA-receptor-associated encephalitis, patients are particularly likely to be young women with ovarian teratomas, presenting with psychiatric or memory disorders, transferred to ICUs with stupor, refractory NCSE, and bizarre orofacial dyskinesias; many respond well to immunosuppressive treatment (and tumor removal) but may take many months to return to normal function (40).
Mimics of NCSE include migraine, transient global amnesia, or sleep disorders, including cataplexy. Others include psychiatric disorders, metabolic derangements, paroxysmal cardiovascular or autonomic dysfunction, endocrine dysfunction, and limbic encephalitis—whether paraneoplastic or related to voltage-gated potassium channel antibodies (39), and confusional states induced by many drugs, such as lithium, other psychotropic medications, or alcohol (24,41).
OVERVIEW OF DIAGNOSIS
While generalized convulsions are usually apparent clinically, nonconvulsive seizures (probably the majority in adults) are harder to recognize. Paradoxically, NCSE may be even harder to recognize than individual seizures because there may be no sudden behavioral change at onset; the patient’s condition may fluctuate little over the day; the significance of many EEG patterns is controversial; and the response to antiepileptic drugs (AEDs) may be subtle or delayed.
The diagnosis of NCSE derives from two major elements: an alteration in baseline cognition, behavior, or other neurologic function, and concurrent epileptiform seizure patterns on the EEG (42,43). Satisfying these criteria is often complicated and difficult. To recognize the clinical features, one must maintain a high index of suspicion. The sign of NCSE is not merely the abnormal behavior, but its change from a baseline state.
To diagnose NCSE, epileptologists have required impaired consciousness for 30 to 60 minutes and an EEG with continuous (44), or at least some form of, seizure activity (42). EEG waveform morphologies may include rhythmic slowing, sharp waves, spikes, and mixtures of these features (45). Discharges may be continuous, persistent with brief pauses of a few seconds, or intermittent. Many EEGs are more ambiguous (see section “EEG Diagnosis of Nonconvulsive Status Epilepticus”), with more blunted waveforms slower than 1.5 Hz, sometimes resembling triphasic waves (TWs) (43,46). Over time, NCSE patterns tend to evolve in morphology, amplitude and frequency, and wax or wane. EEG ictal discharges should be continuous or recurrent for more than 30 minutes, without the patient’s return to a normal clinical state or resumption of the preictal EEG pattern between seizures. Specific EEG features of individual types of NCSE are detailed in the sections below describing those types of NCSE.
Although the EEG is the most reliable diagnostic test for NCSE, an AED challenge may also be useful. Some patients with less “classic” EEG findings show a clinical and electrographic response to BDZs or other AEDs (42). In the appropriate clinical setting, a rapid response to AEDs can be persuasive for a diagnosis of NCSE. Lorazepam may be given in small sequential doses, while monitoring blood pressure, respiratory rate, and oxygenation. To be diagnostic of ongoing seizure activity, there should be prompt improvement in both the patient’s clinical state and the EEG, or complete cessation of electrographic ictal activity with return of normal EEG background activity (43,46) (see Figs. 29.6, 29.14, 29.15, 29.19, and 29.25). Intravenous BDZs may abolish electrographic seizures, but they can also suppress nonepileptic EEG patterns, such as TWs (47), so improvement in the EEG alone upon AED administration does not prove that a particular EEG pattern was a seizure. A controversial EEG pattern is confirmed as ictal only when marked clinical improvement accompanies the EEG improvement—but often such an improvement does not occur. Frequently, the response to AEDs is inconclusive, and the diagnosis must be made on clinical and EEG grounds alone.
Importantly, lack of a prompt improvement with AEDs does not refute a diagnosis of NCSE. Even in patients with definite NCSE, a rapid clinical response is uncommon, especially in obtunded or comatose patients (48). In many, the response is equivocal or substantially delayed (24,49), and sedating BDZs may also impair a clinical response. Following a clinical seizure, if the patient improves on EEG, but not clinically, continuous EEG (C-EEG) monitoring should be considered to look for evidence of subsequent recurrent seizures.
There have also been a few cases of focal NCSE diagnosed clearly by neuroimaging, for example, with 18F-fluorodeoxyglucose (FDG)-positron emission tomography (PET) scans (50,51). Some patients in these reports had clear areas of persistent focal hypermetabolism during clinical episodes of definite aphasia or apparent epileptic amnesia (with an unremarkable EEG, or focal slowing) and an appropriate and quick response to intravenous BDZs. Occasionally, hyperperfusion on ictal single-photon emission computed tomography (SPECT) may help to make, or confirm, the diagnosis (52).
PATHOPHYSIOLOGY
The pathophysiology of convulsive status epilepticus is discussed in Chapter 28. That of NCSE is substantially less well understood, both in the generation of neural activity that produces and sustains NCSE, and in the possibility of its inducing subsequent neuronal damage and longer-lasting epilepsy on its own.
Most NCSE, at least in adults, is either CPSE or secondary generalized NCSE—both with underlying focal onset, whether evident clinically or not. This NCSE usually begins at the same focus of epileptogenesis as for focal seizures that do not become prolonged to the point of SE. How do these focal seizures reach a self-sustaining state?
In a rodent model of limbic SE produced by repetitive, rapid direct electrical stimulation of the hippocampus for 30 to 90 minutes, seizures progress from self-limited to much more prolonged, and eventually self-sustaining SE, persisting for 12 to 24 hours after the stimulation has ceased (53,54). The induced seizures appear similar to those of humans with CPSE— although it is still unclear whether an analogous process occurs in humans. Seizures beginning in the hippocampus appear to promote the propagation of yet more seizure activity, which is maintained in a circuit of structures including the entorhinal cortex, dentate gyrus, and parts of the hippocampus and subiculum (55,56). The initiation of SE in these models was similar to that for individual seizures, but inhibitory or terminating mechanisms failed (see Chapter 28). Generalized seizures, or seizures with onset in other areas, can also precipitate focal temporolimbic seizures and focal SE by way of secondary involvement of the hippocampus or other mesial temporal structures (57).
The activity of certain neurotransmitters can facilitate or inhibit the activation of complex circuits of limbic neuronal activity, prolonging or interrupting nonconvulsive seizures in a (somewhat similar) model of self-sustaining SE produced by stimulation of the perforant pathway in rats (58). For example, injection of galanin (a peptide that inhibits glutamate release presynaptically) into the hippocampus attenuates seizure activity (59). Galanin production can decline during SE, while substance P (which may enhance SE by promoting glutamate release) may increase (60), facilitating the prolongation of seizures. As with GCSE, the balance of excitatory and inhibitory function seems to shift toward excitation as NCSE progresses.
The pathophysiology of absence SE and other forms of primarily generalized SE, however, is starkly different from that of focal-onset NCSE (61). These generalized forms of NCSE involve excessively prolonged synchronization in widespread thalamocortical circuits as part of the generation, maintenance, and reinforcement of aberrant excitatory electrical rhythms in a neuronal system involving both cortical and subcortical structures (55,62). These abnormal electrical rhythms are manifested by rhythmic generalized, anteriorly predominant, spike and polyspike and slow-wave discharges on the EEG.
Experimentally, these 3-Hz generalized cortical spike-and slow-wave discharges can be produced by electrical stimulation of midline intralaminar nuclei of the thalamus (63), and the same thalamic discharges occur during absence seizures in humans (64). The discharges are dependent on low-threshold T-type calcium channel activity (55). Thalamic injections of GABA-B agonists increase the spike-and slow-wave discharges, and this activity can be suppressed by specific AEDs (55).
Little experimental evidence is available on long-term consequences of NCSE. Models of repetitive limbic seizures and SE have been produced by local tissue damage caused by injections of kainic acid, but chemical and electrical methods of inducing SE may damage neurologic tissue independent of the subsequent seizures, that is, it may be the local electrical or chemical precipitant or toxin that damages neurons.
There are a few clinical reports of patients with hippocampal edema during episodes of NCSE, with subsequent hippocampal atrophy in the same area (65,66), but not all studies show this progression to atrophy, possibly depending in part on how prolonged the episodes of NCSE were. These results also contrast with the very minimal clinical morbidity attributable to NCSE directly. Lothman has summarized the many physiologic changes that occur during SE, but most apply primarily to GCSE; some may apply to NCSE (67).
It is very difficult to prove that any neurologic damage accrues from NCSE. Pathologic studies of the effects of GCSE in humans are relatively few, in part because fatal SE cases are often associated with acute, severe brain-injuring illnesses that may cause damage independently. Episodes of NCSE are even less often fatal unless they occur in association with GCSE or other acute, severe neurologic illness—in which case it is difficult to sort out the cause of any neuronal damage. For the most part, pathologic studies of patients with pure forms of NCSE remain unavailable.
The experimental models described in the chapter on GCSE included SE durations of well over 30 minutes (typically 1 to 2 hours of convulsions and electrical discharging activity) to produce neuronal damage in animals. In one rodent model of SE, neurologic damage in the amygdala and pyriform cortex correlated strongly with prolonged “high-frequency” (10 Hz) discharges, but there was no damage following discharges slower than 1 Hz (68). Damage was directly related to the duration and intensity of electrographic seizure activity. It is not clear that such prolonged intense stimulation, or the resultant 10-Hz spike discharges, are an accurate model of human NCSE. Also, in the NCSE model with continuous hippocampal stimulation, neuronal loss did not occur when the interval between nonconvulsive seizures was increased (69). Currently, little is known about the pathologic effects of NCSE in humans. Clinical consequences of NCSE are discussed in later sections.
CLASSIFICATION
No classification system for NCSE will be satisfactory to all basic investigators, electroencephalographers (EEG-ers), clinicians, and scholars. Gastaut stated that there were “as many types of status as there are types of epileptic seizures” (70), but the correspondence is not strict. For example, different focal seizures emanate from different brain areas, with different signs and symptoms, but many can go on to generalized convulsions and GCSE, and to subsequent NCSE after convulsions—with all later stages appearing similar despite the different origins.
Others reject a classification of NCSE according to seizure types, pointing out that epilepsy syndromes such as juvenile myoclonic epilepsy (JME) consist of more than seizures types alone, and also that NCSE is not always simply the prolongation of individual seizures (71). Different NCSE syndromes include EEG, clinical, and genetic bases; developmental aspects; agespecific presentations; varied concomitant structural brain abnormalities or encephalopathies; quite varied responses to treatment; and different longer-term outcome. Thus, NCSE can also be classified based primarily on age and the state of cerebral development or maturation, presence or absence of an encephalopathy, occurrence within an epilepsy syndrome, and, for NCSE of focal origin, understanding by anatomic localization (Table 29.1) (14). This scheme, organized primarily by age of onset, aids in understanding NCSE in the context of the brain’s development and prior condition. Many of the syndromes included occur in neonates, infants, and young children; some continue into adolescence in individuals, or occur in adults.
Boundary syndromes are mostly those with markedly abnormal baseline neurologic conditions and epileptiform abnormalities on EEG—in which it is difficult or impossible to determine what role the epileptiform discharges have in causation of the clinical manifestations. Most are pediatric NCSE syndromes that occur within a broad range of illnesses such as the Lennox-Gastaut syndrome (LGS), and include electrical status epilepticus in sleep (ESES), described briefly here but more in Chapters 25 and 26. Boundary syndromes may also include epileptiform EEG patterns of uncertain significance such as “triphasic” waves or periodic discharges (lateralized or generalized), where there is substantial uncertainty in differentiating whether this represents seizure activity or not (see next section) (41,72). “Subtle” SE often includes prominent myoclonus, usually occurring in seriously ill patients in stupor or coma.
Table 29.1 Classification of Nonconvulsive Status Epilepticus Syndromes
1. Neonatal and infantile syndromes (including):
• West syndrome, Ohtahara syndrome
• Severe myoclonic epilepsy of infancy (SMEI; i.e., Dravet syndrome)
2. Childhood syndromes (including):
• Genetic-based illnesses such as ring chromosome 20 syndrome; Angelman syndrome, Rett syndrome
• Benign childhood epilepsies without a discrete genetic origin, for example, Panayiotopoulos syndrome
• Childhood epileptic encephalopathies such as Landau-Kleffner syndrome and electrical status epilepticus in sleep (ESES) Febrile SE
3. Childhood and adult forms of NCSE:
(a) With epileptic encephalopathy:
(i) For example, within the Lennox-Gastaut syndrome, with atypical absence SE or tonic SE, and the continuation of LGS into adulthood
(ii) Other epileptic encephalopathies
(b) Without epileptic encephalopathy:
• Typical absence SE of idiopathic generalized epilepsies
• Focal NCSE, sometimes referred to as “aura continua” with sensory, autonomic, or cognitive symptoms
• Complex partial SE: limbic or nonlimbic
• NCSE that follows the convulsions of generalized seizures or generalized convulsive SE
• “Subtle” SE with myoclonic SE following convulsions
4. Later adult NCSE: including de novo absence SE of late onset
5. “Boundary syndromes”:
• Some “atypical absence” SE of childhood
• Epileptic behavioral disturbances or psychosis
• Other forms of epileptic encephalopathy and coma due to brain injury (e.g., anoxia) with epileptiform discharges, including “subtle” status.
• Drug or metabolic confusional states with epileptiform discharges (generally not considered true NCSE)
EARLY LIFE, AGE-RELATED NONCONVULSIVE STATUS EPILEPTICUS SYNDROMES
Many neonatal seizures and NCSE have minimal clinical manifestations and remain underdiagnosed if EEG monitoring is not utilized (73,74) (see also Chapter 25), especially in neonates with severe brain injury (75) and in pharmacologically paralyzed infants. Electrographic patterns of neonatal seizures also differ from those of older patients, often remaining localized to relatively small brain areas, likely due to incomplete myelination and neuronal migration (76). Multifocal seizures (more than three foci) are common, especially with multifocal or diffuse brain injury. Some seizures show evolution in frequency, amplitude, morphology, or spatial distribution, but many show little or no such evolution (76). Morphologies include sharply contoured, sinusoidal, or rounded waveforms in the alpha, theta, and delta frequency range, but the frequency and morphology may vary within a single seizure and from seizure to seizure (Fig. 29.1A-C). Many neonatal seizures last 2 to 3 minutes but recur frequently; prolonged continuous seizures are less frequent (77).
Conversely, not all abnormal-appearing neonatal movements are seizures (74). Proper use of EEG and EEG monitoring can help to avoid inappropriate AED treatment of movements such as some swimming or pedaling motions, and stimulus-sensitive clonus or myoclonus, when they are not epileptic in origin. In infants, particular attention must be paid to exclude artifacts mimicking electrographic seizures.
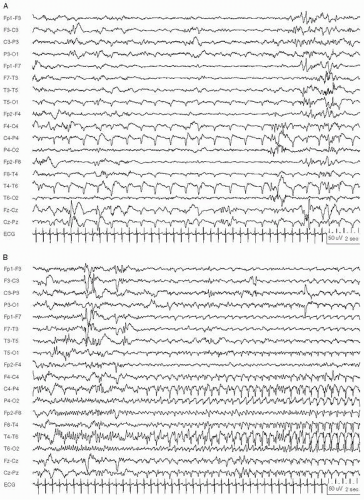
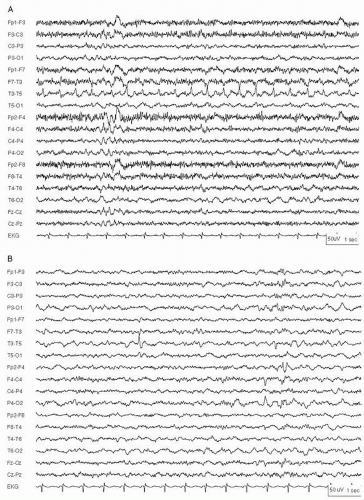
Figure 29.1 Neonatal status epilepticus in a 36-week conceptional age neonate with multifocal infarcts due to a placental abruption following a maternal motor vehicle accident. Multifocal seizures had various electrographic patterns. A: Monomorphic sharply contoured delta activity. B: Continuation of the same seizure, showing evolution into rhythmic spike-and slow-waves at 2.5 Hz, maximal in the right posterior quadrant. C: Later in same seizure, now with rhythmic spike-and-wave over the left hemisphere and independent lower voltage ictal delta activity in the right hemisphere.
Figure 29.1 (Continued)
Several neonatal seizures and SE syndromes have had genetic characterization. Dravet syndrome (severe myoclonic epilepsy of infancy) was originally described as being manifested clinically by prolonged obtundation, referred to as “obtundation status” (78,79). Infantile epileptic encephalopathy, or Ohtahara syndrome, typically presents with greater flexor, extensor, or tonic spasms and prominent suppression bursts on the EEG (80).
Many of the most prominent pediatric NCSE syndromes associated with underlying developmental delay or regression demonstrate activation of epileptiform abnormalities during sleep (81,82). ESES implies activation by sleep of persistent epileptiform activity suggestive of SE, although not all children with these EEGs have clinical seizures. The waking EEG usually shows infrequent focal or occasionally generalized epileptiform discharges, with the spike component usually more prominent than the slow wave. During rapid eye movement (REM) sleep, epileptiform discharges are similar to those on the waking EEG.
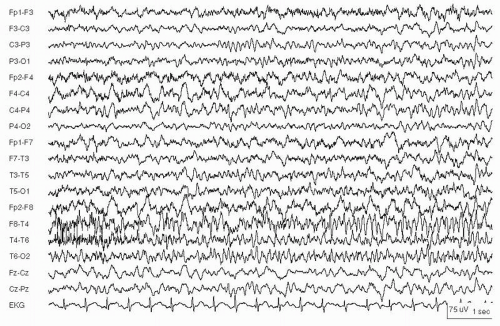
Some children have an associated ESES syndrome, with ESES on the EEG along with progressive cognitive dysfunction, often referred to as an epileptic encephalopathy (83). One such syndrome, continuous spikes and waves during slow sleep (CSWS), is an age-related pediatric epilepsy syndrome, usually starting late in the first decade of life in children who were neurologically normal before seizures began (84). Seizures include generalized convulsions and often evolve to become atypical absence and atonic seizures with dangerous falls (81,85). EEG during wakefulness and REM sleep shows infrequent focal or generalized epileptiform discharges. Non-REM sleep prompts continuous generalized or bilaterally synchronous (if sometimes asymmetric) spike-and-wave complexes at 1.5 to 3.5 Hz (Fig. 29.2). Continuous spike-wave patterns occupying at least 85% of slow-wave sleep are required for the diagnosis of CSWS (Fig. 29.2) (85,86). Most children with CSWS have focal or generalized seizures, global behavioral problems, and regression of cognitive function, more than in language alone (85,87). The epilepsy is often severe and can include many seizure types; some children have prolonged atypical absence SE (81).
CSWS may last for years, but the typical EEG pattern usually occurs between ages 5 and 15 years. The EEG eventually normalizes, including during sleep, and almost all seizures resolve, but cognitive and behavioral problems persist in most cases (81,85). In LGS, the CSWS syndrome may include a prominent component of negative myoclonus or asterixis, at times explaining apparent atonic seizures and dangerous falls (27). These syndromes are covered more extensively in Chapter 26.
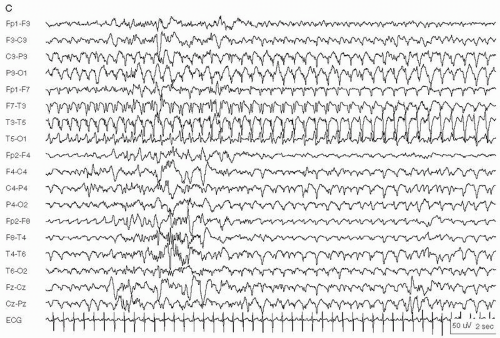
Figure 29.2 Continuous spike waves during slow-wave sleep in a 9-year-old girl with mild developmental delay.
The Landau-Kleffner syndrome (LKS) can be considered another “boundary” syndrome and may overlap with CSWS and other “benign” focal epilepsy syndromes. LKS is characterized by acquired aphasia, seizures, and a behavioral disorder presenting in children with previously normal language development, between the ages of 2 and 4 years (88). A gradual deterioration, specifically in language and speech output, may begin with apparent word deafness or a form of auditory agnosia (89), but there can also be hyperkinetic behavior disorders. LKS exhibits clinical seizures in most cases, though seldom overt SE. It is unclear whether the deterioration in language function is the result of epileptiform activity or if the language deterioration and epileptic activity (both clinical and EEG) result from a common underlying pathologic condition. Most LKS cases are of unknown origin; a few cases are due to symptomatic lesions (90). Corticosteroids are the most common treatment for LKS (91), but most children have persistent language deficits (89,92).
The waking EEG in LKS may be normal or show unilateral or bilateral temporal spike-and-wave discharges. There are sleep-activated bursts of diffuse or multifocal spike-and-wave discharges at 1.5 to 3.5 Hz (ESES), most commonly in anterior and middle temporal regions, but also in temporoparietooccipital regions, some with a very broad, nearly generalized field (Fig. 29.3) (86,93). In contrast to CSWS, epileptiform discharges are present in less than 85% of slow-wave sleep, and may occur during REM sleep (94). The EEG manifestations usually appear between 3 and 5 years of age and resolve at about 15. The EEG may improve over time, with or without treatment (90).
LKS and other ESES syndromes are often treated with AEDs, especially BDZs, with a goal of eliminating epileptiform discharges and the idea (not entirely proved) that the epileptic process contributes to the cognitive deterioration. Although episodes of ESES may recur frequently, the prognosis for seizure control is good, but CSWS has a poor prognosis for cognitive function. LKS and CSWS are rare syndromes, each accounting for about 0.2% of all pediatric epilepsy patients (95).
In ring chromosome 20 syndrome, particularly in patients from Japan between the ages of 13 and 31 years, NCSE consists of prolonged confusional states, with or without motor phenomena, and prominent bilateral or generalized high-voltage slow waves with intermixed spikes on the EEG (96). The epilepsy is characterized by intermittent motor seizures with confusion, staring, head turning, mutism and meaningless utterances, facial flushing, and limb shaking. Impulsive behavior, inappropriate responses, and myoclonus occur in episodes up to several times daily.
Figure 29.3 A 4-year-old boy with language regression, Landau-Kleffner syndrome. EEG during sleep shows bilateral 1-Hz centrotemporal sharp waves, more prominent over the right hemisphere.
Although NCSE in children is perhaps best considered in the context of age and brain development, the descriptions of NCSE in adults in this chapter follow more familiar International League Against Epilepsy (ILAE) seizure types and syndromes, focusing on the type of seizure apparently persisting and causing the patient’s clinical deficit. They are organized into focal and generalized forms, acknowledging that seizures can progress from focal onset to generalized clinical and EEG manifestations. Myoclonic, tonic, and clonic SE are detailed further in the convulsive SE chapter because of their motor manifestations.
FOCAL NONCONVULSIVE STATUS EPILEPTICUS
Focal status epilepticus may be either SPSE with no impairment of consciousness, or CPSE with consciousness impaired. SPSE presents with ictal symptoms reflecting the involvement of discrete regions of brain, such as motor, sensory, special sensory, psychic, or autonomic symptoms (97). Focal SE with motor components is described in Chapter 28. Nonmotor (nonconvulsive) forms of focal SE include persistent sensory disturbances (e.g., visual hallucinations) and autonomic and cognitive deficits, including affective, amnestic, and language disturbances such as aphasia (42).
Overall, focal onset of symptoms and electrographic abnormalities is more common than SE with a generalized onset (98). Focal NCSE is seen frequently after strokes or other acute brain injuries, and should be suspected when patients do not stabilize or improve as expected. In some critically ill patients, NCSE consists of brief cyclical electrographic seizures occurring every 5 to 10 minutes over several hours (99). Focal lesions from vascular causes, trauma, developmental abnormalities (such as heterotopias and other migration abnormalities), and mitochondrial disorders are among many possible causes (100,101).
Focal Sensory Nonconvulsive Status Epilepticus
Persistent sensory seizures (without motor accompaniment) have been called “aura continua” (102,103). Infrequently, they may last for hours. Simple partial sensory status epilepticus is identical to the prolonged continuation of an isolated aura continua. In these cases, local inhibitory function is sufficient to prevent spread of the seizure more broadly in the brain. If, however, these seizures spread to involve areas of the brain necessary for responsiveness (for a long period), this becomes CPSE. Occasionally, benign rolandic epilepsy, in which there is speech arrest, drooling, dysphagia, facial weakness, head deviation, and mild confusion, may be prolonged enough to constitute NCSE (104,105).
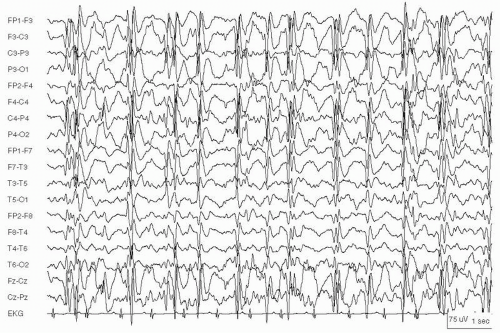
Figure 29.4 An 8-year-old boy with simple partial status epilepticus arising in the right occipital region. Seizures consisted of seeing pulsating green and red circles in the left visual field.
Pure sensory SE is generally considered rare, but this may reflect a detection bias as an EEG may show little at the time of SE (97), and persistent sensory disturbances are often attributed to transient ischemia (106) or other nonepileptic causes. In one report, four patients had focal sensory NCSE lasting up to several days, without motor or behavioral symptoms or alteration in responsiveness (107). Some were frontal or temporal in origin. They appeared to be less common than auras progressing to more obvious seizures, and far less common than CPSE.
Focal lesions in temporal, parietal, and occipital cortices may produce SE with persistent cognitive or sensory symptoms, with hallucinations of any sensation, including visual, auditory, olfactory, gustatory, somatosensory, or vertiginous perception (103). Olfactory hallucinations from temporal epilepsy may be the most common. Some SPSE consist of auditory sensations alone, with epileptiform discharges confined to Heschl’s gyrus (108).
Transient cortical blindness or visual field loss (sometimes referred to as status epilepticus amauroticus) can be a manifestation of occipital SPSE (109), although simple visual hallucinations and ictal blindness are usually brief (110). Occipital seizures can also include macropsia, micropsia, misperception of spatial orientation, hallucinations of faces or animals, or simple patterns of color and light (110) (Figs. 29.4 and 29.5). There may be scotomata or simple visual hallucinations predominating in the central visual field, but complex hallucinations are also possible (111). Slightly more anteriorly, involvement of the temporoparietooccipital junction may produce nystagmus with contraversive eye deviation (20).
The “positive” sensory symptom of pain during SPSE appears even more rare, although shorter painful seizures have been reported (103,112). Some are parietal in origin, in the somatosensory area. Parietal NCSE can also be manifested solely by persistent paresthesias (51). Other unpleasant epileptic sensory seizures may include nausea, ictal fear, anorexia, poorly described visceral sensations, and the distress of “abdominal epilepsy” originating in mesial temporal areas, especially involving the amygdala (112, 113 and 114). Autonomic symptoms are reported increasingly frequently, particularly in children (115).
Some focal SE involve a larger brain area than a typical simple focus, and could be called “regional.” The most common (usually pediatric) form of regional SE is the Panayiotopoulos syndrome, in which an exact focus is often difficult to determine (116). This syndrome is an age-dependent epileptic susceptibility that is usually idiopathic and benign, although perhaps 15% are due to focal lesions. It affects about 13% of all children aged 3 to 6 with nonfebrile seizures. Manifestations are primarily autonomic, with ictal emesis and occasional syncope, usually arising from sleep. Half of the episodes last longer than 30 minutes and thus constitute autonomic SE. Most children have few episodes and typically do better without treatment, and the long-term risk of epilepsy is small (116). Earlier described in correlation with occipital spikes, this syndrome may have discharges elsewhere or multifocally and is probably better considered a regional (but not quite generalized) form of seizures and SE.
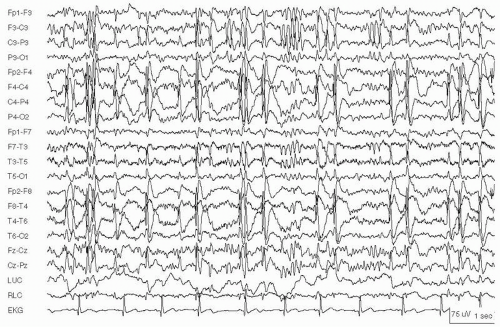
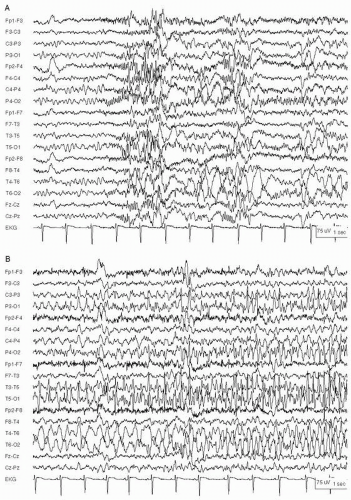
Figure 29.5 Focal status epilepticus in a 14-year-old girl with right occipitoparietal cortical dysplasia. A: Seizure begins with rhythmic beta activity over both occipital regions, more prominent on the right. B: Seizure evolves into high-voltage 3- to 4-Hz spikes in the left occipital region, and spikes with slower frequencies in the right occipital region. Seizures recurred every 2 to 3 minutes for several hours before presentation to the ER.
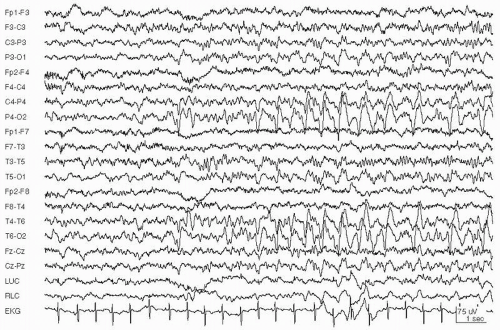
Figure 29.6 A: A 49-year-old woman with mild developmental delay who presented with an anterior aphasia. She was able to follow commands. EEG during aphasia shows repetitive sharp waves in the left temporal region at 2 Hz. B: Same patient after 2 mg IV lorazepam. EEG shows resolution of ictal activity and a left midtemporal spike. The aphasia resolved.
Amnestic and Aphasic Status Epilepticus
Persistent memory dysfunction often follows complex partial seizures in the postictal period, and it may follow, and be due to, an episode of status epilepticus (117,118). Episodes of pure amnesia (with preserved cognitive function in other realms) can also be due to individual epileptic seizures, referred to as “transient epileptic amnesia (TEA)” (119). TEA almost always occurs in patients with earlier epilepsy and is often associated with additional persistent memory deficits. Seizures preferentially affecting memory may require involvement of medial temporal structures bilaterally, as shown by depth electrode recording (120). TEA typically occurs in older patients, thus overlapping clinically, and often difficult to distinguish from (nonepileptic) transient global amnesia. Many episodes of TEA are prolonged beyond 30 minutes, constituting NCSE with amnesia as the sole clinical manifestation (119). Other amnestic syndromes can be caused by frontal or generalized NCSE (121); the frontal discharges suggest an inability to access extant memory during the seizures. Various “psychic” phenomena, including apraxia and acalculia, as well as prolonged mood changes, such as depression and panic attacks, can also occur in NCSE (122).
Aphasic SE, with preserved responsiveness and a clinical deficit restricted to language dysfunction alone, is a focal or regional NCSE (while CPSE implies a disturbance in responsiveness) (123, 124, 125, 126 and 127). Careful language testing is necessary to determine that the deficit is truly an aphasia (125,128). Speech arrest alone can result from seizures in many areas. A focal neurologic deficit in speech production may be mistaken for other syndromes such as transient ischemia or psychiatric illness.
A wide variety of aphasic NCSE types has been characterized by appropriate and thorough language testing—with preserved responsiveness and other cognition except for the aphasia (128). Often, the ongoing epileptiform discharges on EEG correspond anatomically to the area associated with a particular aphasia (Fig. 29.6), such as posterotemporal focal discharges in patients with clinically evident Wernicke’s-type aphasia (124,126). Those with more anterior aphasias (some with frontotemporal discharges on EEG) are often unable to speak but may retain verbal memory and respond appropriately to verbal commands, giving evidence of relatively preserved comprehension (122,123). Some have alexia and reduction in spontaneous speech. Still, the correlation to classical (usually stroke-defined) areas of clinical involvement for different types of aphasia is far from precise (128).
Diagnosis of Focal Nonconvulsive Status Epilepticus
Focal NCSE is usually more subtle in presentation than either focal motor SE or generalized forms of SE. SPSE manifested by sensory or cognitive symptoms alone can be very difficult to diagnose and can be confused with many other illnesses (see Differential Diagnosis above). The EEG is extremely useful in establishing a diagnosis, but not all patients have diagnostic findings. In one report of six patients with prolonged neurocognitive deficits and evidence of focal-onset NCSE, all had rapid rhythmic epileptiform discharges on EEG and eventual resolution of all symptoms with treatment (122).
EEG
Surface EEGs, even when recorded during the symptoms, have limited sensitivity for detection of ictal discharges in SPSE, at least in part because the seizures may involve a small volume of brain tissue. The EEG may be normal or show nonspecific changes (129). One report described 87 well-characterized simple partial seizures in 14 patients, and just 20% of seizures had identifiable epileptiform abnormalities on scalp EEG (97). Two thirds of seizures had primarily sensory symptoms, and these showed ictal EEG changes in just 15%. In another study, only 20% to 35% of simple partial seizures had ictal correlates on surface EEG (130). Sometimes, SPSE can remain undiagnosed until invasive EEG monitoring is performed—usually carried out because of (other) refractory seizures (131,132).
When SPSE is visible on EEG, it usually shows ictal onset with a single topographic EEG maximum, followed by evolution in frequency, voltage, and distribution. Focal fast-frequency discharges, rhythmic waveforms with evolving morphology, repetitive epileptiform discharges, or regional or lateralized alterations in EEG background activities may be seen (129,130). If SE is discontinuous, the background activity between seizures is typically slow and disorganized, with occasional focal interictal epileptiform discharges.
Electrographic focal SE is seen in some stuporous or comatose patients with no clear clinical signs of seizure activity (133). Seizures may be either continuous or repetitive, and EEG patterns are similar to those in SPSE and CPSE. Such partial SE is not rare after strokes or other acute brain injuries and should be suspected when patients in those settings do not stabilize or improve as expected. EEG should be performed for any ICU patient in whom mental status changes are unexplained or appear out of proportion to the degree of acute neurologic injury. Patients with suspected SPSE also warrant an MRI scan to look for an underlying focal lesion.
Complex Partial Status Epilepticus
The first definite case of CPSE was reported by Gastaut and Roger in 1956 (13). In the 1970s, CPSE was considered rare and was the subject of isolated case reports (117,118), and by 1985 only 17 clearly identified cases had been published (134). With the realization that absence SE is a separate category rather than simply any NCSE with generalized discharges, CPSE was eventually recognized as more common (17,18,135), although it remains under-diagnosed (136). Other terms for CPSE include temporal lobe SE (often not an accurate localization), prolonged epileptic fugue state, and psychomotor SE (134,137, 138, 139 and 140).
The definition of SE as a seizure or series of seizures without recovery for 30 minutes (141) allows for the possibility that CPSE can consist of a single very long complex partial seizure or a series of recurrent complex partial seizures without clinical normalization between seizures (Fig. 29.10) (118). CPSE is probably the most common form of NCSE in adults who are ambulatory or not critically ill.
Figure 29.7 “Psychic” status epilepticus in a 41-year-old woman with right mesial temporal sclerosis. She presented with mild confusion but could follow most commands. She did not answer questions and had bizarre speech, repeating “you are my Norm, you are my one true love” continuously.
CPSE begins with a focal-onset seizure, progressing to involve more of the brain such that responsiveness is impaired, and then with prolongation or recurrence. CPSE may last hours, days, or even months (17,142). The most common causes are new acute vascular disease, or old strokes with a more recent precipitant such as infection or metabolic derangement (44,143). Other infections (e.g., encephalitis), tumors, congenital developmental abnormalities, and vascular malformations are possible. The diagnosis of CPSE should be considered in patients who are partially or completely unresponsive, especially those with earlier epilepsy or vascular disease.
While most patients have earlier epilepsy (144), CPSE may arise in patients without prior seizures. CPSE is seldom reported in children, but it may be significantly underrecognized and occur in those with substantial developmental delay and neurologic deficits, and even in those with other seizure types (145). From the incidence of SE in different populations with epilepsy, it has been estimated that CPSE occurs in 35 cases per million population per year, but possibly five times as often in developmentally delayed patients (144).
CPSE may present as abnormal behavior or diminished responsiveness associated with lateralized epileptiform seizure activity. It may begin with a simple partial seizure, or consciousness may be impaired at the onset. Impairment of consciousness, which may cycle or fluctuate, ranges from almost nondiscernible clouding of higher cortical function to coma (44,99,118,145). CPSE may include an “epileptic twilight state” with a lack of responsiveness or confusion, and bizarre, and particularly fluctuating, behavior or paranoia (17,117,118,134,140) (Fig. 29.7). The extensive variety of clinical presentations of CPSE makes EEG essential to distinguish CPSE from other causes of altered mental status.
Lip-smacking, other oroalimentary automatisms, lateralized limb automatisms and dystonic posturing, eye deviation, and nystagmus are typical of CPSE; myoclonus is rare (44,140,146). Fear, aggressivity, irritability and anxiety, and stereotyped, complex automatisms are more frequent with CPSE than with absence SE (146), but when the automatisms or movements are minimal, CPSE can be difficult to distinguish from absence SE.
EEG
In contrast to SPSE, most CPSE shows discernible surface EEG changes. The EEG patterns of CPSE are variable, reflecting differences in the location of ictal onset zones and propagation pathways, but they are often very similar to those seen in isolated complex partial seizures. CPSE may be characterized by either repetitive complex partial seizures, each showing focal onset, with background slowing between seizures, or by continuous ictal rhythms, corresponding to cycling or continuous clinical manifestations (70,99,134,147,148). Rarely, ictal patterns arise independently from both hemispheres (Fig. 29.8). Waveform morphologies include repetitive epileptiform spikes; spike-and-slow wave discharges; rhythmic theta, delta, or alpha frequencies; or rhythmic low-voltage fast activity (45). Occasionally, the EEG can be normal or obscured by artifact (149).
Only gold members can continue reading. Log In or Register to continue