CHAPTER 175 Normal and Abnormal Embryology of the Brain
It is increasingly becoming apparent that although congenital brain and spinal cord malformations share some common clinical features, the morphology, epidemiology, and natural history of these disorders suggest a diverse and heterogeneous embryologic origin.1,2 In our view, a classification of congenital craniospinal malformations based on reputed embryonic mechanisms is much more helpful. We review the normal embryology of the nervous system, as well as the presumed embryonic mechanisms that have been proposed to give rise to various neural tube malformations, with the intent of providing a mechanistic embryonic framework on which to hang these various disorders.
At the outset, it is important to understand that most of the embryologic mechanisms proposed for congenital craniospinal malformations are putative; that is, there is no absolute proof that they are the cause of a particular human malformation, and there are few adequate animal models to test our hypotheses about the origin of human malformations. However, we can gain considerable insight into the embryopathology of congenital malformations through the study of normal neural development. Throughout this chapter we refer to human development in terms of both postovulatory days (PODs) and the embryonic staging system of O’Rahilly and Müller3; the timing and stages for all major developmental events are listed in Table 175-1. Because this chapter reviews only cranial malformations, it does not include discussions specific to spinal cord development or to congenital spinal cord malformations; interested readers are referred elsewhere for this information.4,5
TABLE 175-1 Embryologic Classification of Congenital Craniospinal Malformations
| DISORDERED MIDLINE AXIAL INTEGRATION DURING GASTRULATION |
| LOCALIZED FAILURE OF NEURULATION |
| PREMATURE ECTODERMAL DYSJUNCTION |
| Some spinal lipomyelomeningoceles (cranial to S2) |
| INCOMPLETE ECTODERMAL DYSJUNCTION |
| Dermal sinus tracts, dermoid/epidermoid tumors, ?encephaloceles |
| DISORDERED TELENCEPHALIC CLEAVAGE |
| Holoprosencephaly |
| DISORDERED COMMISSURAL DEVELOPMENT |
| Callosal agenesis, dysgenesis |
| DISORDERED NEURONAL MIGRATION |
| Schizencephaly, lissencephaly, agyria, polymicrogyria, heterotopias |
| DISORDERED RHOMBIC LIP DEVELOPMENT |
| Dandy-Walker malformation, rhombencephalosynapsis |
| POSTNEURULATION DISORDERS |
Normal Early Human Neural Development
Blastogenesis and Gastrulation (PODs 1 to 13)
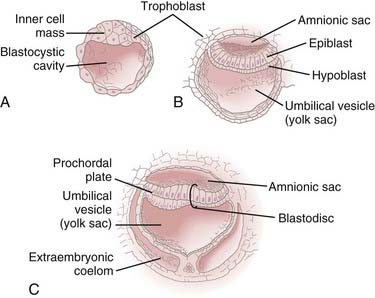
During the first 2 weeks of embryogenesis the human embryo undergoes a number of cell divisions and cellular rearrangements (blastogenesis) that ultimately result in the formation of a blastocyst situated eccentrically within a hollow sphere of trophoblast cells (Fig. 175-1A). The blastocyst later forms a two-layered structure with a dorsal layer, the epiblast, adjacent to the amnionic cavity and a ventral layer, the hypoblast, adjacent to the yolk sac.3 A thickening, the prochordal plate, identifies the cranial end of the embryo (Fig. 175-1B).
Gastrulation begins with the appearance of a midline structure, the primitive streak (PS), at the caudal end of the embryo; the cranial end of the primitive streak is called Hensen’s node. A midline trough, the primitive groove, runs in the midline within the PS; the cranial end of the primitive groove is the primitive pit. The PS elongates cranially over the next 3 days and forms a midline structure in the caudal half of the embryo (Fig. 175-2A). The PS then begins to regress (shorten) back toward the caudal end of the embryo.3 As gastrulation proceeds, cells of the epiblast migrate toward the PS and invaginate through the primitive groove (Fig. 175-2A). The first cells to ingress (while the PS is still elongating) are endodermal cells, which displace the hypoblast cells laterally and form prospective endoderm (the displaced hypoblast cells form extraembryonic tissues).6–10 As the PS regresses, mesodermal cells ingress through the PS between the epiblast and newly formed endoderm and become the mesoderm.9,10 The remaining epiblast cells spread out to replace the cells that have ingressed through the primitive groove and thereby form both the neuroectoderm (the neural tube) and the cutaneous ectoderm (skin). All three germ layers, ectoderm, mesoderm, and endoderm, are derived from the epiblast. Gastrulation transforms the embryo from a two-layered structure to a three-layered structure.11
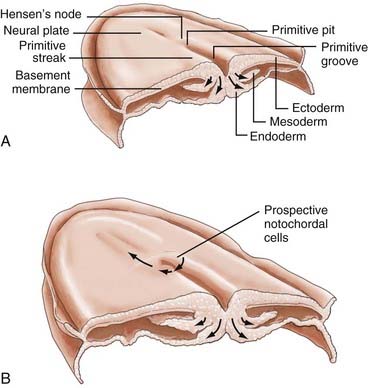
Hensen’s node serves a special role as the “organizer” of the embryo. As the PS elongates, prospective endodermal cells within Hensen’s node migrate through the primitive pit. As the PS regresses, specialized mesodermal cells, prospective notochordal cells, migrate through the primitive pit (Fig. 175-2B) and form the notochordal process in the midline between the neuroectoderm and endoderm.10,11 As we shall see, the notochord plays an important role in directing subsequent neurulation.
To what extent does the notochordal process extend cranially from Hensen’s node, and to what extent does it elongate caudally by the addition of cells to its caudal end from the regressing Hensen’s node? In the chick, the notochordal process grows largely by the latter mechanism.12 In mammals, the available evidence suggests that the situation is much more complex and may involve both mechanisms.13,14 The mechanism of notochord elongation in humans is unknown.
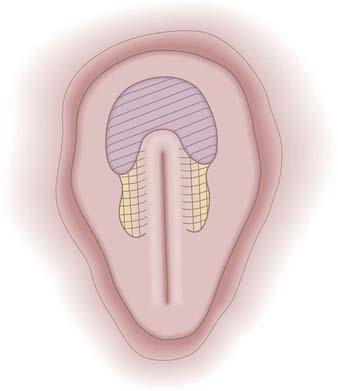
The prospective neuroectoderm is localized to an area of the epiblast that surrounds and flanks Hensen’s node and the cranial half of the PS (Fig. 175-3).15 Fate-mapping studies in chick embryos suggest that each region of the neuroepithelium contributes to multiple neuraxial levels, with the most cranial neuroectoderm contributing to the forebrain and all caudal levels of the neuraxis and more caudal levels of the neuroectoderm contributing to successively more caudal levels of the neuraxis. The prospective brain is derived specifically from neuroectoderm located cranial and lateral to Hensen’s node.15
Formation and Development of the Notochord (PODs 16 to 25)
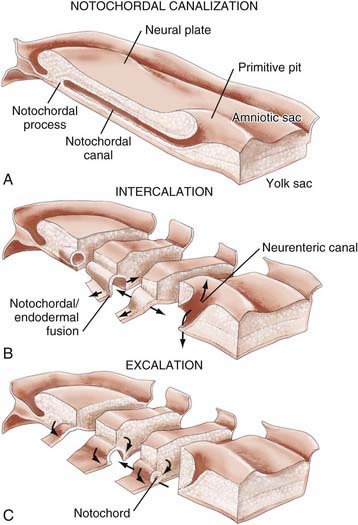
In both human3 and nonhuman primates,16 the notochordal process in cross section consists of cells arranged radially about a central lumen called the notochordal canal (Fig. 175-4A).3 The notochordal canal is continuous dorsally with the amnionic cavity through the primitive pit.17 Between PODs 18 and 20, the notochordal process fuses (intercalates) with the underlying endoderm to form the notochordal plate (Fig. 175-4B). The notochordal plate is therefore incorporated into the roof of the yolk sac, with the notochordal canal becoming continuous with the yolk sac. Intercalation results in a direct communication, the primitive neurenteric canal, that connects the amnionic and yolk sacs at the level of Hensen’s node.3 The neurenteric canal persists for about 3 days, at the end of which the notochordal plate folds dorsoventrally and separates (excalates) from the endoderm (Fig. 175-4C) and the neurenteric canal is obliterated.3,18,19 Thereafter, the true notochord exists as a solid rod of notochordal cells.18 The function or functions of the neurenteric canal are unknown.
Primary Neurulation (PODs 16 to 25)
Human neuroectoderm is first visible at POD 16 as pseudostratified columnar epithelium overlying the midline notochord (Fig. 175-5A) and contiguous with the surrounding squamous epithelium of the cutaneous ectoderm.20 By POD 17 to 19, a midline neural groove (or median hinge point) develops in the midline of the neural plate immediately above the notochord (Fig. 175-5C).21 The neural groove deepens (Fig. 175-5B), and paired neural folds begin to elevate bilaterally.22 Paired dorsolateral grooves (or dorsolateral hinge points) result in further dorsal elevation and medial convergence of the neural folds (Fig. 175-5C). The neural folds separate from the cutaneous ectoderm and fuse (Fig. 175-5D) to form a closed neural tube between POD 21 and 23. Closure generally involves apposition and fusion of first the cutaneous ectoderm and then the neuroectoderm.3,19 The first part of the human neural tube to close is the region of the caudal rhombencephalon or cranial spinal cord, usually when five pairs of somites are present.19
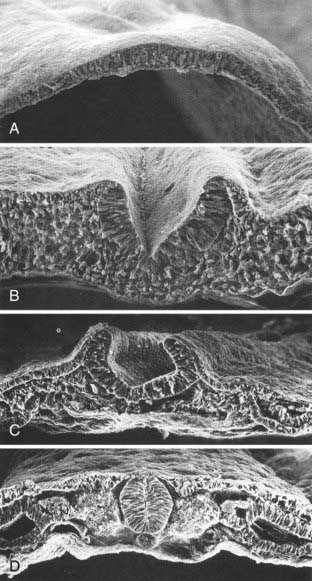
Closure of the neural tube takes place over a period of 4 to 6 days. Although previously thought to close in linear fashion like a zipper extending cranially and caudally from the point of initial closure, mammalian neurulation instead appears to extend from several initiation sites along the craniocaudal neuraxis.19,23–25 Cranial neural tube closure may involve the coordinated interaction of as many as four waves of discontinuous neural tube closure.19,23–26 The spinal cord closes craniocaudally in a linear manner from the point of initial closure to the caudal neuropore.24,25 The last two closure sites are the cranial and caudal neuropores. The cranial neuropore closes between POD 23 and 25, whereas the caudal neuropore closes between POD 25 and 27.27
Neural crest cells are specialized, multipotential migratory cells that arise from the junction between the neural folds and adjacent surface ectoderm18,28–30; in humans, a simultaneous origin for some neural crest cells from the surface ectoderm cannot be excluded.19 Formation of the cranial neural crest begins during elevation of the neural folds before they fuse and continues from the closed neural tube well after the neural folds have fused.18,19,31 The spinal neural crest arises only after closure of the neural tube.3,27
Cranial neural crest cells contribute to the branchial arches (Table 175-2) and the arachnoid and pia mater of the cranium (the dura mater appears to derive from the mesoderm). Spinal neural crest cells undergo terminal differentiation into melanocytes of the body wall and limbs, Schwann cells investing the peripheral nerves, spinal cord meninges, dorsal root and autonomic ganglion cells of the spinal nerves, and adrenal medulla.32 Further details regarding control of migration and terminal differentiation of neural crest cells can be found in several reviews.33–35
TABLE 175-2 Contributions of the Neural Crest to Branchial Clefts
Secondary Neurulation (POD 25)
After caudal neuropore closure at POD 25 to 27, the entire nervous system is covered with cutaneous ectoderm, and more caudal neural development takes place by secondary neurulation.27 This process has been covered in other sources and has no relevance to cranial development; interested readers are referred elsewhere for this information.
Development of Brain Vesicles, Neural Tube Bending, and Formation of Alar and Basal Plate Derivatives (PODs 19 to 40)
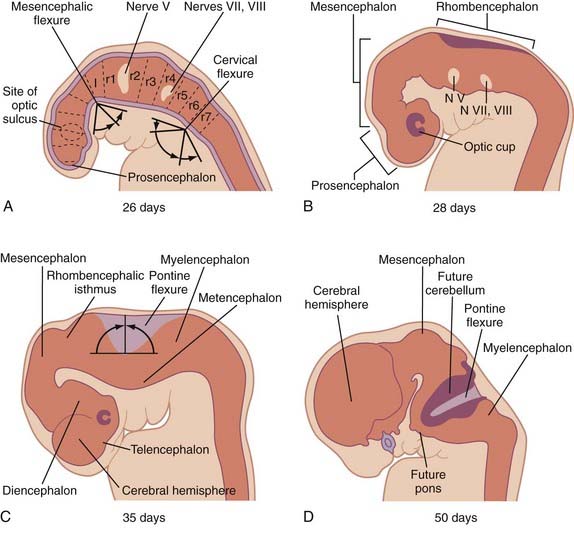
Three neural tube flexures develop during this time as well (Fig. 175-6). The earliest to appear, at POD 19 to 21 during primary neurulation, is the dorsally directed mesencephalic flexure. Later, two additional flexures develop—the dorsally directed cervical flexure (POD 28) and the ventrally directed pontine flexure (POD 32). The pontine flexure continues to bend the future brainstem such that the metencephalon (including the cerebellum) comes to lie dorsal to the myelencephalon by the eighth embryonic week.
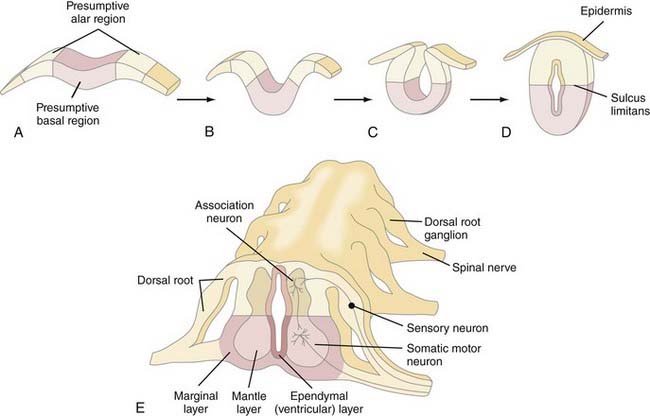
The fundamental cytoarchitectural organization of the spinal cord and brainstem (myelencephalon, metencephalon, and mesencephalon) consists of two components: a basal and an alar plate (Fig. 175-7). The basal plate contains paired ventral neuromotor columns that contribute to the primary motor pathways, whereas the alar plate contains paired dorsal neurosensory columns that contribute to the sensory and integrative pathways. As discussed previously, spinal cord neural crest cells form dorsal root ganglia and dorsal sensory nerves, the central processes of which enter the spinal cord and either ascend within the posterior columns to the nucleus gracilis and cuneatus or synapse locally with dorsal sensory neurons within the alar column (Rexed’s laminae I to VII). Alar plate neurons send projections both cranially in the spinothalamic and spinocerebellar tracts and locally that perform integrative functions with other alar or basal plate neurons within the spinal cord. The basal plate motoneurons receive afferent synapses from alar plate neurons (as well as descending projections from cranial portions of the neuraxis) and, in turn, send efferent axonal projections to the periphery via the ventral root.
The brainstem nuclei are arranged into seven columns, three motor and four sensory. The three motor columns produce somatic efferent, visceral efferent, and special (branchial) efferent nuclei. The four sensory columns produce somatic afferent, visceral afferent, and two special afferent nuclei—special somatic afferent and special visceral afferent nuclei. Each cranial nerve receives contributions from one or more of these seven columns (Table 175-3).
TABLE 175-3 Derivations of Cranial Nerves
| Olfactory (I) | SVA (olfaction) |
| Optic (II) | SSA (vision) |
| Oculomotor (III) | GSE (oculomotor muscles) |
| GVE (ciliary muscle) | |
| Trochlear (IV) | GSE (superior oblique muscle) |
| Trigeminal (V) | GSA (skin, pharynx sensation) |
| GVA (proprioception) | |
| SVE (branchial muscles) | |
| Abducens (VI) | GSE (lateral rectus) |
| Facial (VII) | SVA (taste in the anterior two thirds of the tongue) |
| GSA (skin of the external auditory meatus) | |
| GVA (taste in the anterior two thirds of the tongue) | |
| SVE (muscles of facial expression) | |
| GVE (salivary, lacrimal glands) | |
| Vestibulocochlear (VIII) | SSA (auditory, vestibular) |
| Glossopharyngeal (IX) | SVA (taste in the posterior third of the tongue) |
| GVA (parotid gland, carotid body and sinus, middle ear) | |
| GSA (external ear) | |
| SVE (stylopharyngeus muscle, branchial arch) | |
| GVE (parotid gland) | |
| Vagus (X) | SVA (taste for the palate, epiglottis) |
| GVA (afferents for the trachea, heart, esophagus, stomach, intestines) | |
| GSA (external auditory meatus) | |
| SVE (intrinsic muscles of the pharynx, superior two thirds of the esophagus) | |
| GVE (efferents for the trachea, digestive tract, heart) | |
| Accessory (XI) | SVE (sternomastoid, trapezius) |
| GVE (soft palate, pharynx) | |
| Hypoglossal (XII) | GSE (tongue) |
GSA, general somatic afferent; GSE, general somatic efferent; GVA, general visceral afferent; GVE, general visceral efferent; SSA, special somatic afferent; SVA, special visceral afferent; SVE, special visceral efferent.
Neuronal Proliferation, Neuronal Migration, and Formation of Commissures (PODs 24 to 115)
The cellular architecture of the brain and spinal cord is based on a similar unifying theme—embryonic neuroblasts proliferate within a ventricular zone and their postmitotic progeny migrate centripetally through the neural tube to take up locations peripherally to form the mantle layer. Axonal processes from mantle layer neurons extend further to form the marginal zone immediately under the pia (Fig. 175-8). Neuronal proliferation begins on POD 24; neuroblasts are later replaced by glioblasts at the ventricular zone, which then give rise to glial cells (astrocytes and oligodendroglia). The remaining cells of the ventricular zone become ependymal cells.
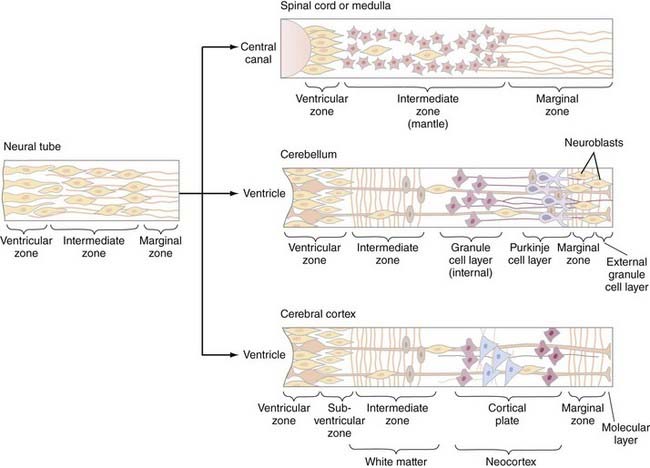
Spinal cord development follows this basic theme and gives rise to the central location of the spinal cord gray matter with surrounding white matter tracts. However, proliferation and migration at other levels of the neuraxis are altered depending on the specific location—two locations that deserve special discussion are the cerebellum and cerebral cortex. In the cerebellum, the postmitotic neuroblasts migrate centripetally to form two zones: the deep cerebellar nuclear layer (which will form the deep cerebellar nuclei) and the Purkinje cell layer (to which the Purkinje, basket, stellate, and Golgi cells migrate) (Fig. 175-8, middle). The granule cells, in contrast, originate in the rhombic lip, migrate superficially around the surface of the cerebellum, and exist superficial to the Purkinje cell layer as the external granule layer. These cells are still mitotically active and produce numerous granule cell progeny, even as long as 2 years after birth.36 Postmitotic granule cells migrate back toward the ventricular zone, through the Purkinje cell layer, to produce the internal granule layer. This second migration is controlled by specialized glial cells, called Bergman glia, the processes of which serve as guideposts for the migration of granule cells through the Purkinje cell layer.
Neurogenesis and neuronal migration are also modified in the cerebral cortex (Fig. 175-8, bottom). The first neuroblasts send axon that form the marginal zone immediately subjacent to the pia. Postmitotic neurons then migrate away from the ventricular zone to form the intermediate zone between the ventricular and marginal zones. Cells from both the ventricular and intermediate zones migrate further centripetally to form the cortical mantle zone—cells that arrive at the mantle zone first form the deeper cortical layers, whereas those that arrive later form more superficial cortical layers that migrate through the previously established layers. As ventricular zone proliferation ceases, proliferation begins in the subventricular zone, located between the ventricular and intermediate zones. Cells from the subventricular zone migrate through the intermediate zone to form a subplate just beneath the cortical mantle zone. Cells from the subplate zone migrate through the mantle zone to form the most superficial cortical layers. The cortex is therefore derived from both the cortical mantle zone and the subplate region and is generated in an inside-out fashion, with superficial layers forming later than deeper layers. The intermediate layer subsequently forms the white matter of the cerebrum. Neuronal migration in the cerebrum also appears to involve specialized radial glial fibers that span from the ventricular to the marginal zones and along which neurons migrate to their final destinations. Neuronal migration involves the interplay of many different molecular species, including reelin, astrotactin, integrins, neurogulins, and others.36
The majority of the commissural fibers of the hemispheres develop from the commissural zone of the lamina terminalis. The lamina terminalis is the site of the cranial neuropore and is located immediately dorsal to the optic chiasm anatomically. Commissural fibers of the anterior commissure, corpus callosum, and hippocampal commissure derive from the dorsal end of the lamina terminalis (the posterior and habenular commissures are formed from the epithalamus of the diencephalon). The earliest commissural fibers, those of the anterior commissure, arise at 54 days, followed by those of the hippocampal commissure during the 11th week and those of the corpus callosum between 84 and 115 days.37,38 The corpus callosum generally develops in an anterior-to-posterior fashion (genu first, followed by the body and then the splenium); because of the later growth of the frontal lobes, the rostrum is the last portion to form. As a result, incomplete abnormalities of the corpus callosum involve the later developing, more posterior portions (and rostrum) with sparing of the earlier developing anterior portions and lead to selective enlargement of the atrium and occipital horns of the ventricles (colpocephaly). An exception to this rule is holoprosencephaly, in which there is selective agenesis of the posterior portions with sparing of the anterior portions.38,39
Abnormal Early Neural Development
Failure of Neural Tube Closure—Anencephaly and Myelomeningocele
Anencephaly and myelomeningocele are neural tube defects (NTDs), or localized failure of primary neurulation, that can arise through one of two mechanisms. The “nonclosure theory” proposes that NTDs represent primary failure of neural tube closure.40 The “overdistention theory,” introduced in 1769 by Morgagni41 and popularized by Gardner,42–45 proposes that NTDs arise through overdistention and rupture of a previously closed neural tube. The nonclosure theory is widely accepted for most cases. However, overdistention may contribute to some experimental NTD models, particularly those caused by vitamin A46 and the T-curtailed mouse mutant47; whether this causes human malformations is unknown. The result is an open, unneurulated segment of neural tube, the nature and severity of which are determined by the location and length of the unneurulated segment. Failure of caudal neurulation produces a myelomeningocele, whereas more cranial failure results in anencephaly.
Because neural tube closure requires the complex interaction of multiple cellular processes, it is not surprising that NTDs may result from a number of embryonic insults. NTDs have been produced experimentally with a number of teratogens (reviewed by Campbell and colleagues2), genetic mutations (reviewed by Copp and associates47,48), and experimental manipulations (reviewed by Schoenwolf and Smith49). Although these processes all suggest a number of potential mechanisms whereby NTDs might arise, the cause of human malformations remains unknown. NTDs are most likely etiologically heterogeneous1,2,47,48 and represent the end result of a variety of embryonic disorders.
Genetic models of NTDs provide a means of identifying candidate gene or genes involved in neural tube closure47,48; three such examples illustrate the variety of ways in which genetic mutations can result in an NTD. The splotch (Sp) mouse mutant is identified by a peculiar patch of white fur and exhibits both exencephaly and myelomeningocele. The genetic locus for the Sp mutation is within the Pax-3 gene locus. Although the Pax-3 gene product appears to be involved in apposition and fusion of the neural folds, neither the mechanisms underlying the normal Pax-3 gene product nor the mechanisms whereby the Sp mutation alters Pax-3 function are known with certainty.
A second mouse mutant, curly tail (ct), exhibits posterior NTDs and tail deformities associated with delay in closure of the caudal neuropore.48 However, the delay is not the result of faulty neuroepithelial development inasmuch as isolated neuroepithelium from ct mutants undergoes normal neurulation.50 Rather, a delay in cell proliferation in the underlying notochord and hindgut endoderm causes an abnormal ventral body axis curvature and impedes posterior neuropore closure.47,48 If the ventral curvature is corrected by splinting the caudal ct embryo with an eyelash51 or by retarding neuroepithelial proliferation with retinoic acid,52 the posterior neuropore closes normally and the incidence of NTDs is reduced.
A third mouse mutant, T-curtailed (T[c]) produces a lumbosacral myelomeningocele with dorsoventral forking of the caudal neural tube. However, rather than delayed or failed neural tube closure, the T(c) mutant causes rupture of the roof plate and reopening of a previously closed neural tube.47
A large number of environmental causes of NTDs have been identified and their underlying mechanisms studied. Recent attention has focused on the role of folate in the embryogenesis of NTDs. Maternal administration of folate antagonists such as aminopterin has long been known to produce NTDs.53 Periconceptional administration of supplemental folate in randomized, placebo-controlled studies reduced both the recurrence rate of NTDs in women with a previously affected pregnancy and the incidence in women who had never had an affected pregnancy.54,55 However, studies of maternal serum and red blood cell folate levels in mothers of infants with myelomeningocele have produced inconsistent results (reviewed by Wald56 and Seller57), and folate deficiency does not cause NTDs in mouse or rat embryos (reviewed by Fleming and Copp58). These observations suggest that NTDs are not usually the result of an absolute folate deficiency.
More recent attention has focused on the possibility that NTDs are caused by abnormalities involving metabolic pathways (in either the mother or fetus) that require folate59; such abnormalities predispose an individual to NTDs and might therefore be overcome by folate supplementation. Folate and its metabolites tetrahydrofolate and 5-methyltetrahydrofolate are important in a variety of mammalian metabolic reactions, including purine and pyrimidine (and therefore DNA) synthesis, and in the transfer of methyl groups during the metabolism of methionine and homocysteine. In particular, the role of folate in methionine and homocysteine metabolism has generated considerable interest.60–68
One hypothesis is that maternal or fetal mutations in either methionine synthase or 5,10-methylenetetrahydrofolate reductase may slow this “methylation cycle” down, drive the conversion of methionine to homocysteine, and lead to methionine deficiency or homocysteine excess, or both. Ingestion of higher doses of folate may overcome this relative deficiency by restoring more normal homocysteine and methionine levels.59,67,69 A number of observations support this view. For example, elevated maternal serum68 and amnionic fluid67 homocysteine levels have been identified in women carrying a fetus with an NTD. Disordered methionine metabolism has been demonstrated in nonpregnant women who had previously given birth to a child with an NTD.67,69 Finally, mutations of the 5,10-methylenetetrahydrofolate reductase gene have been demonstrated in 18% of individuals with NTDs, in 13% of parents of children with NTDs, and in just 6% in controls.65,70 Abnormalities of cystathionine synthase have also been identified.67
A number of teratogens are known to act through a variety of cellular mechanisms to cause NTDs in animals and humans (see Copp and coworkers47 for review). One important teratogen, valproic acid (VPA), produces NTDs in both animal models and humans, probably by inhibiting neural fold fusion.47 Although the exact mechanism is not known, VPA appears to disrupt folate metabolic pathways,71 perhaps by interfering with the conversion of tetrahydrofolate to 5-formyltetrahydrofolate.67 Maternal folate administration has reduced the incidence of VPA-associated NTDs in some, but not all studies,71 and mouse strains that are susceptible to valproate-induced NTDs have significantly lower levels of 5,10-methylenetetrahydrofolate after valproate administration than do resistant strains.72 In addition, a number of developmental regulatory genes (such as transcription factors and cell cycle checkpoint genes) may be altered in these susceptible strains.73 One hypothesis is that VPA may act by changing folate-dependent methylation of regulatory proteins such as transcription factors.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


