CHAPTER 139 Overview of Skull Base Tumors
Classification
A practical classification of skull base tumors divides them into benign and malignant neoplasms. Meningiomas, pituitary tumors, and vestibular schwannomas are the most common benign neoplasms of the skull base, whereas chordomas and chondrosarcomas are the most common malignant ones. In addition to harboring primary tumors, the skull base may be a metastatic site for tumor originating elsewhere in the body; prostate cancer and breast cancer are the most common cancers that metastasize to the skull base, followed by lung cancer and lymphoma.1 Moreover, the skull base may be invaded, most often in continuity, by malignant neoplasms originating in the sinonasal tract (including esthesioneuroblastoma), in the nasopharynx (squamous and non–squamous cell carcinoma), in the oropharynx, or in the ear region. In addition, tumors originating in the orbit may invade the skull base, as is the case with rhabdomyosarcoma, a common malignant intraorbital tumor of children, or benign tumors such as orbital meningioma and osteoma, found mainly in adults. In this chapter we present an overview of these tumors and their clinical findings and briefly discuss approaches to their treatment.
Epidemiology
As just stated, meningiomas, pituitary tumors, and vestibular schwannomas are the most common benign skull base tumors.2 Because about 40% to 50% of meningiomas involve the skull base3 and the clinical incidence of meningiomas is 4.5 per 100,000 per year,2 the incidence of skull base meningiomas would be about 2 per 100,000 per year. The incidence of pituitary tumor is about 1 per 100,000 per year,2 and that of vestibular schwannomas is similar.2,4 Data on skull base metastases are limited. One study quotes a 4% occurrence of skull base metastases in cancer patients, which would give an incidence of skull base metastases of about 18 per 100,000 per year.1
Meningiomas
Meningiomas are the most common primary skull base tumors and are thought to originate from arachnoidal cap cells, cells forming the outer lining of the arachnoidal membrane. The female-to-male ratio is 2.2 : 1, and they constitute 32% of brain tumors.2 Although meningiomas are generally considered to be cured after total resection including their dural attachment, such is not always the case. Indeed, the 5-year recurrence rate for a totally removed World Health Organization benign (grade I) meningioma is 5%, and this risk increases to 40% for totally removed atypical (grade II) meningiomas and to 50% to 80% for malignant (grade III) meningiomas.5 Clearly, this classification has management implications insofar as the use of postoperative radiation therapy is concerned. Luckily, grade II or grade III meningiomas rarely involve the skull base.6 The clinical manifestations of skull base meningiomas have been described in detail.7 Suffice it to say that meningiomas become symptomatic at a smaller or larger volume, depending on their location; cavernous sinus, clival, or meningiomas centered at the internal auditory canal or at Meckel’s cave become symptomatic, in general, at a smaller volume than do anterior cranial fossa, petroclival, or posterior pyramid meningiomas growing posteriorly from the internal auditory meatus.
Therapeutic Plan
Once a meningioma is discovered, it behooves the physician, usually a neurosurgeon, to formulate the most appropriate therapeutic plan by taking into account the patient’s symptoms, age, and general medical condition and the location of the tumor and its size, in addition to an objective self-assessment of the surgeon’s own experience. Moreover, an essential element of this decision must take into account the fact that meningiomas respond to radiation therapy, as has been shown multiple times in the 1970s and 1980s.8 Our capacity to deliver safe radiation therapy has certainly increased in the 1990s and the first decade of the 21st century because of our ability to deliver stereotactic radiosurgery/radiotherapy in a very conformal fashion by using, among others techniques, Gamma Knife radiosurgery, intensity-modulated radiation therapy, tomotherapy, CyberKnife radiosurgery, and proton beam therapy. The combination of better delivery and better imaging has resulted in a tumor control rate of about 90%.9 This better delivery, in conjunction with our better imaging capabilities, has resulted in plans that are able to concentrate a clinically effective dose of radiation on the target while keeping the amount of radiation on structures at risk at a minimum. Radiosurgery delivered in single or multiple fractions must be considered when deciding the best treatment option for a patient with a meningioma, whether as an alternative or as an adjuvant to surgery. Radiosurgery must be regarded as a neurosurgical tool, and as such the neurosurgeon must have a general understanding of what it can do, as well as its limitations. Our philosophy is to offer microsurgical removal to all symptomatic patients in whom we believe we can safely remove the tumor; however, we also present to these patients the option of radiosurgery in single or multiple fractions. The consideration for single or fractionated radiosurgery depends on the tumor’s volume, shape, and proximity to vital structures, among other issues. We may perform surgery with the knowledge that by design we are not going to remove the whole tumor, such as for tumors invading the cavernous sinus. On other occasions we modify our goal of total removal based on circumstances encountered during surgery, such as invasion of neurovascular structures by the tumor or tight pial adhesions in vital areas such as the brainstem or hypothalamus. In all cases in which tumor removal is subtotal, we recommend postoperative radiosurgery whenever the meningioma is atypical or anaplastic and generally monitor benign meningiomas with frequent MRI to detect growth, and only then do we recommend radiosurgery.10,11 Treatment of an asymptomatic patient with an incidentally discovered tumor is strongly affected by patient’s age and preferences, tumor location and size, and the patient’s comorbid conditions.11 Although the data available do not support any clear-cut recommendations, we are more inclined to advise treatment, again microsurgical or radiosurgical, for younger patients and observation and repeated scans for older patients.
Surgical Considerations
 We routinely use a fused computed tomography (CT)-MRI data set for navigation purposes. The bone CT data set is particularly useful when extensive bone removal is needed, such as in the case of any approach involving traversal of the middle cranial fossa or drilling of the pyramid. This data set allows precise identification of middle fossa and pyramidal structures, such as the carotid canal, petrous pyramid ridge, and labyrinthine structures.12 We routinely use the microscopic focus point as the navigation pointer (Fig. 139-1 and Video 139-1).
We routinely use a fused computed tomography (CT)-MRI data set for navigation purposes. The bone CT data set is particularly useful when extensive bone removal is needed, such as in the case of any approach involving traversal of the middle cranial fossa or drilling of the pyramid. This data set allows precise identification of middle fossa and pyramidal structures, such as the carotid canal, petrous pyramid ridge, and labyrinthine structures.12 We routinely use the microscopic focus point as the navigation pointer (Fig. 139-1 and Video 139-1).
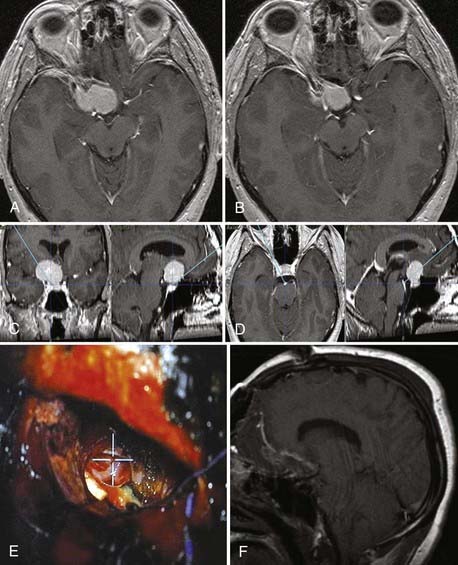
 We use computed tomography angiography (CTA) or magnetic resonance angiography (MRA), or both, whenever the tumor is closely related to major arterial vessels or abuts/involves major dural sinuses (Fig. 139-2 and Video 139-2). Conventional angiography is used whenever CTA/MRA findings are doubtful or when we want to perform a trial occlusion on major vessels. In the event that conventional angiography is performed, we request embolization of any major feeder, but it is uncommon that we resort to angiography for the sole purpose of embolization unless the tumor appears overly vascular on other tests, such as might be the case with hemangiopericytoma. Indeed, it is uncommon for embolization to add any significant value to surgery on skull base meningiomas.
We use computed tomography angiography (CTA) or magnetic resonance angiography (MRA), or both, whenever the tumor is closely related to major arterial vessels or abuts/involves major dural sinuses (Fig. 139-2 and Video 139-2). Conventional angiography is used whenever CTA/MRA findings are doubtful or when we want to perform a trial occlusion on major vessels. In the event that conventional angiography is performed, we request embolization of any major feeder, but it is uncommon that we resort to angiography for the sole purpose of embolization unless the tumor appears overly vascular on other tests, such as might be the case with hemangiopericytoma. Indeed, it is uncommon for embolization to add any significant value to surgery on skull base meningiomas.
Pituitary Adenomas
Pituitary adenomas have a yearly incidence of about 1 per 100,0002 and account for about 10% to 15% of intracranial tumors.13 Their incidence is the same for women and men, and they are more commonly discovered between the third and sixth decades of life, with the median age at diagnosis being 49 years.2
Pituitary adenomas can be functioning or nonfunctioning (hormonally active or nonactive) and can be microadenomas (≤10 mm) or macroadenomas (>10 mm). Symptoms depend on the combination of tumor size and secreting or nonsecreting status. Usually, nonsecreting tumors become symptomatic because of impaired vision caused by compression of the optic apparatus, whereas functional tumors become symptomatic as a result of hormone-related symptoms and signs. In addition to symptoms and signs caused by hormone deficiency or excess and by tumor size, pituitary apoplexy develops in 2% to 7% of pituitary tumors at some point in their course or as the initial symptom.14 Pituitary apoplexy is the result of hemorrhage in the tumor and can cause severe headache (95% to 100%), acute visual deterioration (52% to 64%), oculomotor palsy (78%), and decreased level of consciousness (8%).15 It may be triggered by any type of surgery, pregnancy, anticoagulation, or pituitary hormonal stimulation. Apoplexy may develop in any pituitary adenoma, irrespective of its size or type.16 Seventy percent of pituitary tumors are hormonally active.13,16 Indeed, 97% of microadenomas and 70% of macroadenomas are secretory, and 90% of nonsecreting tumors and only 35% of secreting tumors are large. Approximately 63% of adenomas are macroadenomas.15
Therapeutic Plan
Prolactin-secreting tumors are the most common pituitary adenomas and represent about 50% of them.17 Untreated, prolactinomas are dangerous not only because of the depressed pituitary function and visual compromise associated with a large tumor but also because of osteoporosis associated with increased prolactin levels.18 Patients with prolactin-secreting tumors are typically offered medical treatment with dopamine agonists, with surgery being reserved for those who are resistant to or intolerant of medical treatment. Surgery may be offered as a first option when visual deterioration has occurred rapidly. Radiosurgery may be offered when patients fail both medical management and surgery. Cure is defined as normalization of the prolactin serum level to less than 25 ng/mL in females and less than 20 ng/mL in males, even though some authors use a level of less than 10 ng/mL on the first postoperative day.19 Lowering of prolactin levels after dopamine agonist therapy occurs within days, but tumor shrinkage can take months. Dopamine agonists are successful in normalizing prolactin levels in more than 90% of patients.18 About 10% to 20% of microadenomas and 20% to 30% of macroadenomas are resistant to dopamine agonists, and 5% to 10% of patients are unable to tolerate dopamine agonists.20,21 Transsphenoidal removal of prolactinomas is associated with a cure rate of about 90% with microadenomas18 and less than 50% with macroadenomas, but cure is elusive with tumors invading the cavernous sinus.18 Recurrence after initial normalization occurs in about 20% of patients.22 Radiation therapy is the last choice for pharmacologically and surgically resistant adenomas; cure as defined by a prolactin level of less than 30 ng/mL was reported by Pan and colleagues in 50% of 128 patients with a mean tumor diameter of 13 mm treated by Gamma Knife radiosurgery.23
Abnormal growth hormone secretion is associated with up to a twofold increase in mortality in comparison to the general population secondary to cardiovascular, respiratory, and malignant sequelae of this condition.24 Surgery is the treatment of choice and achieves about a 90% biochemical cure rate (normal insulin-like growth factor type I [IGF-I] level) for microadenomas, a 50% cure rate for macroadenomas (67% cure rate for noninvasive macroadenomas), and less than a 6% recurrence rate after initial biochemical cure.24 A combination of medical therapy with dopamine agonists or somatostatin analogues, or both, and radiosurgery, either in single or multiple fractions, is considered for patients who fail to achieve biochemical cure.
Witt reviewed 20 radiosurgery studies published between 1997 and 2002; cure rates (normalization of IGF-I) were mostly in the 20% to 30% range.25
Untreated Cushing’s syndrome is associated with a 5-year mortality of 50%.26 However, any continuous excess of cortisol may alter glycemic control and blood pressure and may therefore be associated with decreased life expectancy.27 Surgery is the treatment of choice for adrenocorticotropic hormone (ACTH)-secreting tumors (Cushing’s disease). Cure is defined as a postoperative cortisol level of less than 1 µg/dL, and these patients generally require glucocorticoid replacement for up to 1 year.24 As with all pituitary tumors, microadenomas have a greater chance of cure (>90%) than do macroadenomas (≈50%) or invasive adenomas (≈20%).28 When surgery fails, radiosurgery may be used and achieves cure rates of about 50%.25 Treatment with adrenal enzyme inhibitors may have a role in decreasing cortisol levels before surgery or waiting for the effect of radiosurgery. Bilateral adrenalectomy is occasionally performed when surgery or radiosurgery fails.
Thyroid-stimulating hormone (TSH)-secreting pituitary tumors are rare and represent less than 1% of pituitary tumors. Almost all of them are macroadenomas. Surgery is the treatment of choice, and radiosurgery needs to be considered if endocrinologic cure is not achieved. In one study, octreotide normalized TSH levels in up to 79% of patients.24
Nonfunctioning pituitary adenomas are the second most common pituitary adenoma after prolactinoma and represent about 20% of all pituitary adenomas; 80% of them are of gonadotroph cell origin.17,29 The treatment of choice is surgical removal. In a recent study, complete surgical removal was possible in 64% of 491 patients with nonfunctioning pituitary adenomas (all, with the exception of one, were macroadenomas).30 Patients with clean MRI findings postoperatively had a 5-year recurrence-free survival rate of 87%, whereas patients with residual tumor on MRI had a 5-year recurrence-free survival rate of 100% when they received prophylactic radiation therapy and only 68% when they did not.30 The use of radiation therapy postoperatively is associated with a 30% to 70% rate of pituitary insufficiency.31,32
Surgical Considerations
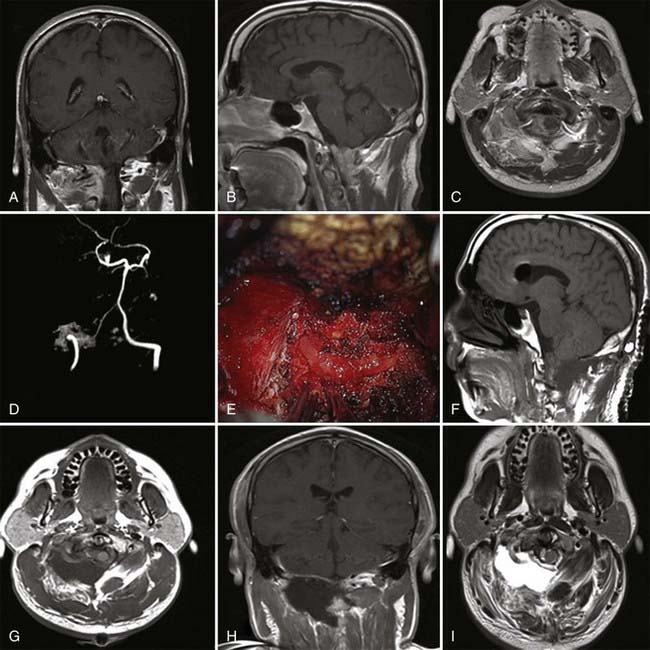
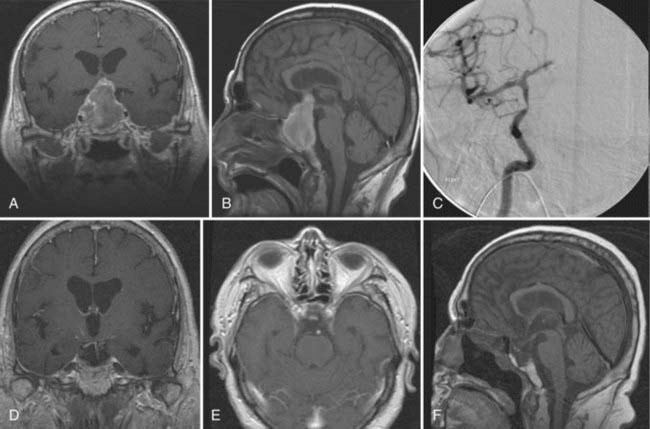
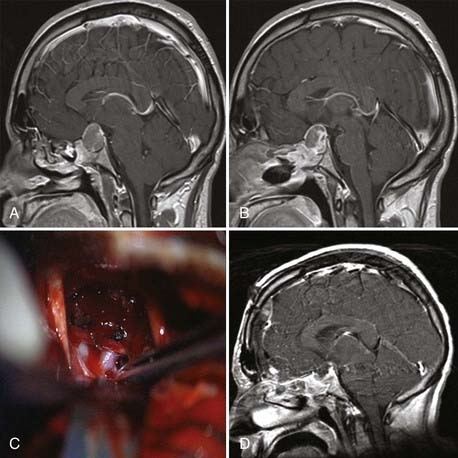
 As pointed out by Ciric and colleagues, there are three basic surgical principles that when strictly adhered to help minimize the complication of transsphenoidal microsurgery while increasing its effectiveness.33 First, the pituitary gland is an extra-arachnoidal structure; the diaphragma sellae arachnoid (suprasellar arachnoid) separates the gland and its tumors from the neurovascular structures contained in the subarachnoid space, such as the optic nerves, the chiasm, the anterior communicating artery, and others. Even in macroadenomas there is always an arachnoidal layer, albeit stretched, separating the gland from the subarachnoid space. If one performs surgical maneuvers outside this layer, complications such as leakage of cerebrospinal fluid (CSF) and neurovascular injury are minimized. Visualization of intra-arachnoidal structures is dangerous. Second, transsphenoidal pituitary microsurgery must be performed as close as possible to the sagittal plane; straying from the sagittal plane puts lateral structures, such as the intracavernous carotid arteries, at jeopardy. Third, the anterior pituitary is always anterior to the tumor, despite being stretched thin by a macroadenoma. Moreover, transsphenoidal microsurgery is an intrasellar operation (excluding the extended transsphenoidal approach), which means that suprasellar tumor extensions are removable insofar as they descend into the sellar space and are more difficult to remove in fibrous tumors, which tend to remain stuck in the suprasellar space. The microsurgical approach to the sella is well engrained in the neurosurgical armamentarium. This is supported by the fact that its complications are minimal even when done by neurosurgeons who do not perform a great number of these procedures.34 The microsurgical approach to the sella has evolved in the past 3 decades from an ororhinoseptal to a rhinoseptal and lately to a direct transnasal approach. The evolution of this approach has been associated with a decreased amount of mucosal dissection and diminished need for postoperative packing of the nasal passages (Fig. 139-3).33 Endoscopic approaches to the sella have been developed and refined in the past 10 years. There are variations in the implementation of endoscopic approaches to the sella, such as uni-nostril versus bi-nostril,12,35 endoscope holder versus no endoscope holder, and turbinate removal versus no turbinate removal.36 The technique that is most popular and that gives the most operative freedom is the bi-nostril approach with at least unilateral resection of the middle turbinate and two surgeons, one holding the endoscope and the other operating through both nostrils. The advantages of the endoscopic approach to the sella seem to reside in better illumination, in the ability to widely expose the inside of the sphenoid sinus, and in more freedom of surgical trajectories inside the sella.12,37–42 Its disadvantages are the lack of tridimensional vision and increased otolaryngologic morbidity. From a clinical point of view, the results seem to be equivalent to those of pituitary surgery but with probably an increased CSF leak rate during the learning curve for the endoscopic approach. Of course, long-term clinical results are available for microsurgical sellar approaches but not for endoscopic approaches. We use both approaches interchangeably and are often prompted to use the endoscopic approach by patients’ requests for what is perceived to be a minimally invasive approach. We also do not hesitate to use extended transsphenoidal approaches for huge pituitary tumors (Fig. 139-4) or transcranial approaches for fibrous tumors that we believe are unable to be removed safely through a transsphenoidal approach (Fig. 139-5 and Video 139-3).
As pointed out by Ciric and colleagues, there are three basic surgical principles that when strictly adhered to help minimize the complication of transsphenoidal microsurgery while increasing its effectiveness.33 First, the pituitary gland is an extra-arachnoidal structure; the diaphragma sellae arachnoid (suprasellar arachnoid) separates the gland and its tumors from the neurovascular structures contained in the subarachnoid space, such as the optic nerves, the chiasm, the anterior communicating artery, and others. Even in macroadenomas there is always an arachnoidal layer, albeit stretched, separating the gland from the subarachnoid space. If one performs surgical maneuvers outside this layer, complications such as leakage of cerebrospinal fluid (CSF) and neurovascular injury are minimized. Visualization of intra-arachnoidal structures is dangerous. Second, transsphenoidal pituitary microsurgery must be performed as close as possible to the sagittal plane; straying from the sagittal plane puts lateral structures, such as the intracavernous carotid arteries, at jeopardy. Third, the anterior pituitary is always anterior to the tumor, despite being stretched thin by a macroadenoma. Moreover, transsphenoidal microsurgery is an intrasellar operation (excluding the extended transsphenoidal approach), which means that suprasellar tumor extensions are removable insofar as they descend into the sellar space and are more difficult to remove in fibrous tumors, which tend to remain stuck in the suprasellar space. The microsurgical approach to the sella is well engrained in the neurosurgical armamentarium. This is supported by the fact that its complications are minimal even when done by neurosurgeons who do not perform a great number of these procedures.34 The microsurgical approach to the sella has evolved in the past 3 decades from an ororhinoseptal to a rhinoseptal and lately to a direct transnasal approach. The evolution of this approach has been associated with a decreased amount of mucosal dissection and diminished need for postoperative packing of the nasal passages (Fig. 139-3).33 Endoscopic approaches to the sella have been developed and refined in the past 10 years. There are variations in the implementation of endoscopic approaches to the sella, such as uni-nostril versus bi-nostril,12,35 endoscope holder versus no endoscope holder, and turbinate removal versus no turbinate removal.36 The technique that is most popular and that gives the most operative freedom is the bi-nostril approach with at least unilateral resection of the middle turbinate and two surgeons, one holding the endoscope and the other operating through both nostrils. The advantages of the endoscopic approach to the sella seem to reside in better illumination, in the ability to widely expose the inside of the sphenoid sinus, and in more freedom of surgical trajectories inside the sella.12,37–42 Its disadvantages are the lack of tridimensional vision and increased otolaryngologic morbidity. From a clinical point of view, the results seem to be equivalent to those of pituitary surgery but with probably an increased CSF leak rate during the learning curve for the endoscopic approach. Of course, long-term clinical results are available for microsurgical sellar approaches but not for endoscopic approaches. We use both approaches interchangeably and are often prompted to use the endoscopic approach by patients’ requests for what is perceived to be a minimally invasive approach. We also do not hesitate to use extended transsphenoidal approaches for huge pituitary tumors (Fig. 139-4) or transcranial approaches for fibrous tumors that we believe are unable to be removed safely through a transsphenoidal approach (Fig. 139-5 and Video 139-3).
Craniopharyngiomas
Craniopharyngiomas are benign extra-axial, usually intra-arachnoidal neoplasms. They are thought to originate either from rests of the craniopharyngeal duct (the connection between the oral cavity and Rathke’s pouch, the evagination of the oral cavity that will form the adenohypophysis) or from squamous metaplasia of the developing pituitary. There are two histologic subtypes of craniopharyngioma, classic adamantinomatous tumors, which represent up to 95% of pediatric cases,43 and the papillary squamous epithelium subtype, which is mainly found in adults.44 Craniopharyngiomas typically display a combination of cystic, solid, and calcified components. Their incidence is about 0.1 per 100,000 per year, and they account for less than 1% of all primary brain tumors, with an equal distribution between males and females.2 Craniopharyngiomas characteristically have two age peaks, one at 5 to 14 and the other at 50 to 74 years.44 Although they are found in more than 90% of children with sellar-suprasellar pathology,45 50% of craniopharyngiomas are nonetheless diagnosed in adults.44 A minority of craniopharyngiomas (about 10%) are solely intrasellar, and even fewer are located exclusively inside the third ventricle. The majority of craniopharyngiomas (>80%) have their bulk located in the suprasellar area.43
Headache, nausea, and vomiting are more common in children, probably because hydrocephalus is more frequent in the pediatric population. All other symptoms are equally common in adults and children.44 Weeks to years may elapse between onset of the first symptoms and diagnosis, both in children and in adults.44
At diagnosis, endocrine dysfunction is found in up to 80% of pediatric patients; reduced growth hormone secretion is present in up to 75% of pediatric patients, followed by FSH/LH deficiency (40% of patients) and ACTH and TSH deficiency (25% of pediatric patients).44 According to some studies the rate of demonstrable endocrinologic abnormalities is the same in children and adults.44
Therapeutic Plan
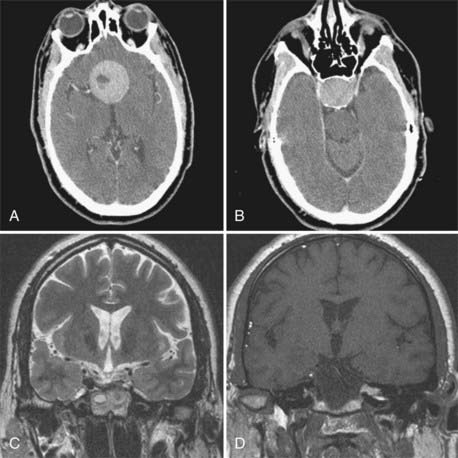
Treatment must be individualized according to tumor characteristics (balance among cystic, solid, and calcified portions), patients’ age and symptoms, and availability of resources. Although it is clear that the best treatment of a benign neoplasm is total removal, this needs to be tempered by the fact that anatomically, craniopharyngiomas are at times very tightly adherent to the floor of the third ventricle, they occasionally grow in true subpial fashion, and aggressive removal of them may be accompanied by significant neurological deficits. Four treatment modalities are effective in managing these tumors: microsurgery, intracystic irradiation, intracystic chemotherapy, and focused beam radiation (Gamma Knife or linear accelerator based).46 For nonrecurrent tumor with a considerable solid component, microsurgery with the goal of complete resection and avoidance of iatrogenic morbidity, even at the expense of total resection, is reasonable. Indeed, total resection is rarely achieved when long follow-up is available.46 Cystic components may be well managed by drainage through an Ommaya reservoir and intracavitary instillation of antibiotic or radioisotopes (Fig. 139-6). Solid areas of postsurgical residual tumor may be well controlled with focused beam radiation if symptomatic or if growth is demonstrated. Surgery for recurrent tumors is usually associated with significant complications and a low chance of achieving total tumor removal. With the use of multimodality treatment, a great number of tumors may be controlled with maintenance of good quality of life. Because of the biologic behavior of these tumors, surveillance is a lifelong event, with multiple therapeutic modalities being used at different treatment points. Again, it is mandatory for the neurosurgeon to be conversant with all the available treatment modalities to be able to customize treatment and intervene at the appropriate decision-making points.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


