▪ PATHOPHYSIOLOGY
To better understand the clinical approach to diagnosis and evaluation of pain syndromes, the student must understand the basics of pain physiology. It is only with a solid grasp of this knowledge that a rational approach to pain pharmacology can be made.
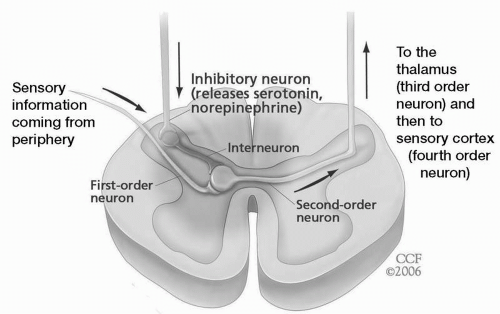
In the classic neospinothalamic pathway, the first-order neuron, with its nociceptors, transduces and carries impulses to the dorsal horn of the spinal cord (or its counterpart in the brainstem, the trigeminal nucleus caudalis), where it synapses with the second-order neuron. In turn, this second-order nociceptive neuron travels up the spinal cord on the opposite side to synapse with a third-order neuron in the thalamus. From there the third-order neuron travels through the thalamocortical pathway to bring pain to the primary sensory and association cortices. Superimposed on the synaptic connections at the dorsal horn is a descending antinociceptive or analgesic pathway arising from the brainstem (periaqueductal gray, locus ceruleus, and raphe nuclei) and dependent on the monoamines, norepinephrine, and serotonin.
At each synaptic junction, there are interneurons capable of inhibiting the synaptic connection (via gamma-aminobutyric acid [GABA]) or enhancing it (through inhibition of inhibitory interneurons). Each neuron depends on depolarization at the membrane level and relies on voltage-gated sodium and calcium channels that respond to neuropeptides secreted by communicating neurons. Conduction along each neuron in turn depends on the functional integrity of the axon and the insulation surrounding it (
Fig. 25.1).
Damage anywhere along this system induces both positive (e.g., paresthesias, increased appreciation of pain, and spontaneous jolts of pain) and negative sensory symptoms (numbness and sensory loss). However, the nociceptive system, unlike other sensory systems in the body, exhibits positive phenomena with afferent sensory damage, which likely emanate from remaining intact synaptic connections on hyperactive brainstem or spinal neurons (see the upper two neurons in
Fig. 25.1). Additionally, interference with or damage to the descending
antinociceptive modulatory pathways can lead to enhancement of pain-conducting traffic along the pathways. This enhancement is dependent on excitatory amino acids, specifically glutamate, secreted by the first-order neuron into the synapse. Glutamate binds to receptors on the second-order nociceptive neuron, opening up the voltage-gated calcium channel and resulting in depolarization (and excitability). Such hyperexcitability is known as sensitization of the second-order neuron and is associated with the phenomena of temporal summation of pain, allodynia, spontaneous pain, and causalgia (burning pain).
Two pain mechanisms are known to be involved in the excitability of neurons, making them more sensitive to stimuli and sensory inputs—peripheral and central sensitization.
Peripheral Sensitization (first order neuronal activation)
Nociception occurs in the context of a number of changes in the chemical environment brought about by local tissue damage and the release of various inflammatory mediators. In the process of peripheral sensitization, some of these mediators act to enhance the sensation of pain in response to stimuli in the affected area by decreasing stimulus thresholds and increasing and prolonging nociceptors’ firing and intensity of response to suprathreshold stimuli. Prostaglandin E2, released from arachidonic acid-damaged cells, binds to G protein-coupled prostaglandin receptors to sensitize nociceptors. Interleukin-1b and tumor necrosis factor-α, released by immune system cells involved in the inflammatory response induce the release of cyclooxygenase-2 (COX-2) several hours after the start of inflammation, and COX-2 converts arachidonic acid to prostaglandin H and eventually prostaglandin E2. Prostaglandin E, bradykinin, nerve growth factor, and leukotrienes also directly sensitize the nociceptors.
Certain peptides released by primary afferent neurons themselves support the process of peripheral sensitization. Substance P, neurokinin A, and calcitonin gene-related peptide (GCRP) released from activated nociceptors act on local mast cells to increase histamine release, further amplifying the process in neurogenic inflammation. Peripheral sensitization is thought to be one of the processes underlying the phenomenon of primary hyperalgesia, whereby injury to peripheral tissues results in an enhanced sensation of pain in response to subsequent suprathreshold painful stimuli. The evolutionary purpose of hyperalgesia is to discourage further contact with damaged tissue, thereby expediting the healing process.
Peripheral sensitization and the consequent hyperalgesia normally resolve as tissue heals. However, chronic pain may occur if the body is unable to restore homeostasis because the initial injury has exceeded the body’s capacity for recovery or the injury involves the nervous system itself.
Secondary hyperalgesia—the expansion of hyperalgesia beyond the region of initial tissue damage—is caused by additional nervous system changes secondary to the initial insult and is further described below.
Central Sensitization (second order neuronal activation)
The pathogenesis of central sensitization may be thought of as occurring in two phases. The first stage is triggered by intense nociceptive input to the secondary afferent neurons of the dorsal horn and may thus be considered activity dependent. This excessive input may arise from persistent acute injury, surgical insult, and peripheral sensitization of nociceptors during inflammation, ectopic discharge from nerve injury, a variety of chronic pain syndromes, or other conditions. The activity-dependent stage of central sensitization occurs rapidly; hyperre-sponsiveness of spinal neurons may be seen within seconds of massive sensory input triggered by an appropriate initial insult in the periphery.
N-Methyl-D-aspartate (NMDA) receptors on secondary afferent neurons of the dorsal horn (or the trigeminal nucleus caudalis in the brainstem) are of critical importance in the pathology of central sensitization. Activation of NMDA receptors by glutamate leads to a number of signaling cascades initiated by increased intracellular calcium influx, activation of G protein-coupled neurokinin receptors, tyrosine kinases, and phosphokinases, leading to phosphorylation of NMDA receptor ion channels and a decrease in the magnesium block of these channels. This release from magnesium blockade acts to functionally open the calcium channel to the influx of extracellular calcium and depolarizes the neuron. As a result, sensitized (depolarized) second-order afferent neurons respond more avidly to continued C fiber afferent input from the periphery; spontaneous or ectopic signal discharge may occur in the context of this loss of gatekeeping function. At some point, the second-order nociceptive neurons in the dorsal horn or trigeminal nucleus caudalis no longer abide by activity-dependent behavior (primary afferent input) and start to exhibit spontaneous activity. To the unfortunate patient, this is interpreted as sudden shooting or tic-like pain, causalgia (unprovoked burning pain), and the appreciation of nonnoxious stimuli as painful (allodynia). In the latter instance, second-order dorsal horn neurons that normally respond to tactile stimuli (wide dynamic neurons) interpret tactile stimuli inside and outside their normal receptive fields as noxious.
Classification of Pain
There are several ways to classify pain. Some specialists arbitrarily separate pain into malignant (cancer-related) and nonmalignant (non-cancer-related) pain. In the daily clinical practice of pain medicine, the experienced practitioner often resorts to a functional classification of pain into two categories: nociceptive (somatic and visceral pain) and nonnociceptive (neuropathic and idiopathic pain).
Somatic pain is caused by the activation of pain receptors in either the cutaneous tissues (body surface) or deep tissues (musculoskeletal tissues). Deep somatic pain is usually described as dull or aching but localized. Surface somatic pain is usually sharper, may have a burning or pricking quality, and is aggravated by movement. Common causes include postsurgical pain and pain related to soft tissue trauma.
The deep somatic nociceptors associated with muscles, tendons, fascia, joints, and periosteum are largely free nerve endings with A delta and C fibers. The afferent fibers carrying pain impulses enter the spinal cord through the ventral and, especially, the dorsal roots. The lightly myelinated A delta fibers terminate primarily in laminae I and V of the dorsal horn of the spinal cord. Unmyelinated, slowly conducting C fibers terminate in laminae II. More rapidly
conducting sensory afferents enter the spinal cord by way of the substantia gelatinosa and terminate in lamina I and IV.
Visceral pain results from stimulation of pain receptors in the wall of viscera, and these are sensitive to mechanical distention. Most visceral nociceptors are unmyelinated, slowly conducting polymodal C fibers. In fact, one can burn and cut visceral structures, such as the gut, without inducing pain, but cannot distend it without causing intense discomfort. The pain is described in different terms from somatic pain; it is diffuse, achy, cramping or spasmodic, and generally poorly localized. Visceral pain often is accompanied by the perception of pain arising in somatic structures that share the same spinal level of innervation. This phenomenon, known as referred pain, results from the convergence of nociceptive fibers from different structures onto the same population of cells in the dorsal horn. This explains such recognizable phenomena as pain associated with gastric perforation that causes both upper gastric aching pain and left shoulder pain.
Neuropathic pain is the result of damage or disfunction occurring anywhere along the neuraxis—the peripheral nerve, spinal ganglion, nerve root, spinal cord, brainstem, thalamus, and cortex, but the damage may not be readily detectable at the bedside. The words used by patients to describe the pain help distinguish it from the nociceptive pain types mentioned earlier and incorporate descriptors for both positive and negative sensations—burning, searing or scalding, cold, numb, tingling, shooting, stabbing, crushing, and vice-like. These sensations, which affect not only the sensory system, but also the affective, cognitive, and attention systems, result from the disruption of the body’s normal pain-conducting circuitry. A thorough discussion of the theories underlying the generation of neuropathic pain is beyond the scope of this article. Interested readers are referred to recent detailed reviews on the subject.
Neuropathic pain can result from abnormal discharges in injured or regenerating sensory afferents. Injured nerve fibers sometimes develop spontaneous discharges, and afferent impulses may be evoked by electrical activity in surrounding axons in injured nerves. A variety of mechanisms are involved in pain accompanying nerve disease, and multiple mechanisms may be operating simultaneously in any given patient. These mechanisms include the following:
Spontaneous discharges in injured nociceptive afferents
Induced discharges in injured nociceptors afferents
Deafferentation with subsequent changes in dorsal horn or central nociceptive transmission (i.e., loss of inhibitory interneuronal input), leading to central pain generation
Activation of sensitized mechanoreceptors by efferent sympathetic activity
Specific ion channel changes in primary afferents
Finally, idiopathic pain has been used interchangeably with the term psychogenic pain. Idiopathic pain is the more appropriate term because it implies a wider spectrum of poorly understood pain states. The Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM-IV) refers to “pain disorder associated with psychological factors (acute and chronic)” and “pain disorder associated with both psychological factors and a general medical condition (acute and chronic).” This term has begun to replace the older terminology of psychogenic pain in instances in which no identifiable organic origin could be found. Fibromyalgia, regional myofascial pain, and somatoform pain are examples of idiopathic pain. In addition to the lack of organic origin, the pain and associated symptoms often are thought to be grossly out of proportion to any identifiable disorder.