Chapter 4 Parkinsonism
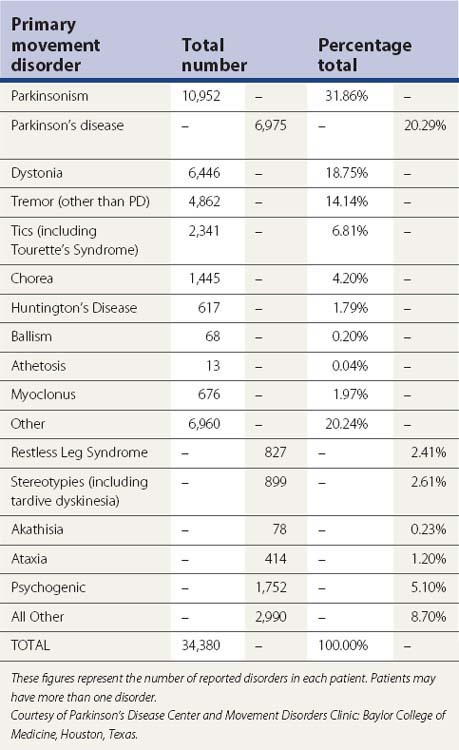
Clinical features and differential diagnosis
Introduction
Parkinsonism is a syndrome manifested by a combination of the following six cardinal features: tremor-at-rest, rigidity, bradykinesia, loss of postural reflexes, flexed posture, and freezing (motor blocks). A combination of these signs is used to clinically define definite, probable, and possible parkinsonism (Table 4.1). The most common form of parkinsonism is the idiopathic variety known as Parkinson disease (PD), first recognized as a unique clinical entity by James Parkinson in 1817, who in his An Essay on the Shaking Palsy identified six cases, three of whom he personally examined and the others he observed on the streets of London (Parkinson, 1817). Previously referred to as “paralysis agitans,” Charcot later in the nineteenth century gave credit to Parkinson by referring to the disease as “maladie de Parkinson” and pointed out that slowness of movement should be distinguished from weakness; he also recognized non-tremulous forms of PD (Kempster et al., 2007). With the recognition of marked clinical-pathologic heterogeneity of parkinsonism due to a single mutation and some uncertainty whether PD should be defined clinically, pathologically, or genetically, a variety of other names have been proposed for this neurodegenerative disorder, including “Parkinson complex” and “Parkinson Lewy disease” (Langston, 2006), but it is unlikely that these names will replace the traditional name “Parkinson disease.” Some have argued that PD is not a single entity, a notion supported by genetic forms of parkinsonism with variable clinical and pathologic features (Weiner, 2008).
Table 4.1 Parkinsonism diagnostic criteria
It was not until 100 years after Parkinson’s landmark paper that the loss of dopamine-containing cells in the substantia nigra (SN) was recognized (Tretiakoff, 1919). In 1960, Ehringer and Hornykiewicz (1960) first noted that the striatum of patients with PD was deficient in dopamine, and the following year, Birkmayer and Hornykiewicz (1961) injected levodopa in 20 patients with PD and postencephalitic parkinsonism and noted marked improvement in akinesia but not in rigidity. Later in the same decade, Cotzias and colleagues (1967, 1969) are credited with making levodopa clinically useful in patients with PD. The recent disclosure of the diagnosis of PD in several public figures has contributed to increased awareness about the disease, which should translate into greater research funding.
Clinical features
There are dozens of symptoms and signs associated with PD, and the clinician must become skilled in eliciting the appropriate history and targeting the neurologic examination in a way that will bring out and document the various neurologic signs (Jankovic and Lang, 2008; Tolosa et al., 2006; Jankovic, 2007, 2008). The manifestation of PD may vary from a barely perceptible tremor to a severe disability during the end-stage of the disease. Rest tremor in the hands or in the lips might be not just socially embarrassing but may cause a severe handicap in people whose occupation depends on a normal appearance. Therefore, it is important that the severity of the disease be objectively assessed in the context of the individual’s goals and needs. In some cases, unintended movements accompanying voluntary activity in homologous muscles on the opposite side of the body, the so-called mirror movements, may occur even in early, asymmetric PD (Espay et al., 2005; Li et al., 2007), although one study showed that mirror movements actually occur less frequently in PD patients than in healthy controls (29% vs. 71%, P < 0.0001) (Ottaviani et al., 2008).
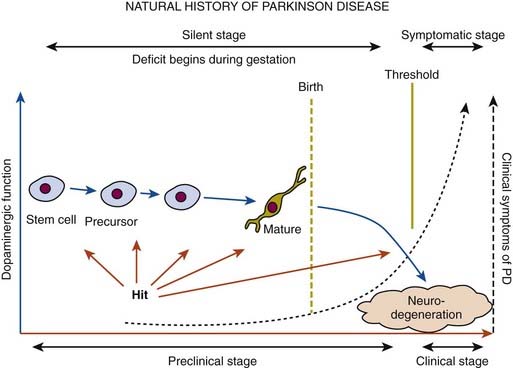
In a retrospective study of patients with PD, early nonspecific symptoms that were reported included generalized stiffness, pain or paresthesias of the limbs, constipation, sleeplessness, and reduction in volume of the voice (Przuntek, 1992). More specific complaints that were elicited on a detailed history as the disease progressed included problems with fine motor skills, decreased sense of smell, loss of appetite, and a tremor occurring with anxiety. Family members retrospectively reported decreased arm swing on the affected side, decreased emotional expression, and personality changes, including more introversion and inflexibility. Using strict criteria for asymmetry, 46% of patients with PD had characteristic asymmetric presentation that correlated with handedness (Uitti et al., 2005). Handedness, however, did not predict the onset of PD motor symptoms in another study (Stochl et al., 2009). The mechanisms of the observed asymmetry of PD symptoms and signs are not well understood, but the side of predominant involvement may be stochastically determined, similar to other complex diseases such as cancer (Djaldetti et al., 2006). The notion that the side of predominant involvement is merely coincidental and determined by chance alone is supported by seemingly random right and left distribution without correlation to hand dominance, lack of concordance for the affected side within family members of genetically determined PD, and the frequent presence of asymmetric involvement in drug-induced parkinsonism. In some cases, parkinsonism may remain confined to one side and may be associated with hemiatrophy. In one study, the mean age at onset of the 30 patients who satisfied the criteria for hemiparkinsonism–hemiatrophy was 44.2 (15–63) years with a mean duration of symptoms of 9.7 (2–20) years (Wijemanne and Jankovic, 2007) (Table 4.2). Half of all patients had dystonia at onset and dystonia was present in 21 (70%) of all patients during the course of the syndrome. The majority of patients were responsive to levodopa, and perinatal and early childhood cerebral injury appeared to play an important role in about half of the cases. This syndrome of hemiparkinsonism–hemiatrophy also suggests that some cases of PD may start prenatally, and as a result of the low number of dopaminergic neurons from birth and subsequent age-related attrition, develop PD symptoms in middle age (Le et al., 2009) (Fig. 4.1). The asymmetrical lateral ventricular enlargement that is associated with PD motor asymmetry (Lewis et al., 2009) may represent a nonspecific marker of underlying neurodegeneration or may suggest an early insult, similar to what has been postulated in hemiparkinsonism–hemiatrophy (Wijemanne and Jankovic, 2007).
Clinical heterogeneity of PD and the rich phenomenology associated with the disease are well recognized. In a survey of 181 treated PD patients, Bulpitt and colleagues (1985) found at least 45 different symptoms that were attributable to the disease, nine of which were reported by the patients with more than fivefold excess compared with a control population of patients randomly selected from a general practice. These common symptoms included being frozen or rooted to a spot, grimacing, jerking of the arms and legs, shaking hands, clumsy hands, salivation, poor concentration, severe apprehension, and hallucinations. Hallucinations, although usually attributed to dopaminergic therapy, may be part of PD, particularly when there is a coexistent dementia and depression (Fenelon et al., 2006; Marsh et al., 2006). However, even these frequent symptoms are relatively nonspecific and do not clearly differentiate PD patients from diseased controls. Gonera and colleagues (1997) found that 4–6 years prior to the onset of classic PD symptoms, patients experience a prodromal phase characterized by more frequent visits to general practitioners and specialists in comparison to normal controls. During this period, PD patients, compared to normal controls, had a higher frequency of mood disorder, fibromyalgia, and various pains (Defazio et al., 2008), particularly shoulder pain (Stamey et al., 2008; Madden and Hall, 2010). In one study of 25 PD patients and 25 controls, PD patients had 21 times the odds of having shoulder pain compared with those without PD (Madden and Hall, 2010).
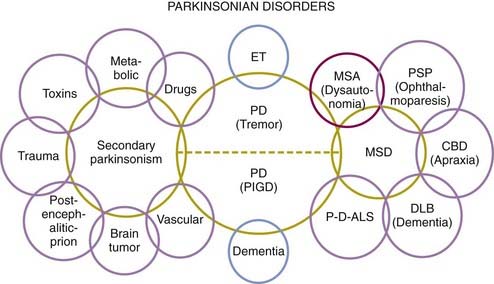
Because of the marked heterogeneity of clinical symptoms and natural progression, several studies have attempted to identify clinical subtypes of PD. Using cluster analysis, a systematic review of the literature confirmed the existence of the following disease subtypes: (1) young age at onset and slow disease progression, (2) old age at onset and rapid disease progression, (3) tremor-dominant, and (4) postural instability and gait difficulty (PIGD) (Jankovic et al., 1990) dominated by bradykinesia and rigidity (van Rooden et al., 2009). Patients who manifest predominantly axial symptoms, such as dysarthria (Ho et al., 1999), dysphagia, loss of equilibrium, and freezing of gait, are particularly disabled by their disease in comparison to those who have predominantly limb manifestations (Jankovic et al., 1990; Muslimovic et al., 2008). The poor prognosis of patients in whom axial symptoms predominate, many of whom have either the PIGD form of PD or some atypical parkinsonism (Fig. 4.2), is partly due to a lack of response of these symptoms to dopaminergic drugs (Jankovic et al., 1990; Kompoliti et al., 2000). In this regard, one study suggested that an abnormal tandem gait (the “ten steps” test) is much more common in patients with atypical parkinsonism than in those with typical PD and this test seems to differentiate the two groups with 82% sensitivity and 92% specificity (Abdo et al., 2006). In another classification of PD subtypes, the differences are largely driven by the severity of “nondopaminergic” features and levodopa-related motor complications (van Rooden et al., 2011).
Bradykinesia
Bradykinesia, the most characteristic clinical hallmark of PD, may be initially manifested by slowness in activities of daily living and slow movement and reaction times (Cooper et al., 1994; Touge et al., 1995; Giovannoni et al., 1999; Jankovic et al., 1999a; Rodriguez-Oroz et al., 2009). In addition to whole-body slowness, bradykinesia is often manifested by impairment of fine motor movement, demonstrated on examination by slowness in rapid alternating movements. Although speed and amplitude are usually assessed together on the Unified Parkinson’s Disease Rating Scale (UPDRS) Part III, there is some evidence that amplitude is disproportionately more affected than speed in patients with PD and may be due to different motor mechanisms and should probably be assessed separately (Espay et al., 2009). Other manifestations of bradykinesia include drooling due to failure to swallow saliva (Bagheri et al., 1999; Lal and Hotaling, 2006), monotonic and hypophonic dysarthria, loss of facial expression (hypomimia), and reduced arm swing when walking (loss of automatic movement). Micrographia has been postulated to result from an abnormal response due to reduced motor output or weakness of agonist force coupled with distortions in visual feedback (Teulings et al., 2002). Bradyphrenia refers to slowness of thought. Bradykinesia, like other parkinsonian symptoms, is dependent on the emotional state of the patient. With a sudden surge of emotional energy, the immobile patient may catch a ball or make other fast movements. This curious phenomenon, called kinesia paradoxica, demonstrates that the motor programs are intact in PD but that patients have difficulty utilizing or accessing the programs without the help of an external trigger. Therefore, parkinsonian patients are able to make use of prior information to perform an automatic or preprogrammed movement, but they cannot use this information to initiate or select a movement. Although PD represents the most common form of parkinsonism, there are many other causes of bradykinesia, the parkinsonian clinical hallmark (Table 4.2).
The pathophysiology of bradykinesia is not well understood, but it is thought to result from failure of basal ganglia output to reinforce the cortical mechanisms that prepare and execute the commands to move (Jankovic, 2007). This is manifested by slowness of self-paced movements and prolongation of reaction and movement time. Evarts and colleagues (1981) first showed that both reaction (RT) and movement (MT) times are independently impaired in PD. The RT is influenced not only by the degree of motor impairment but also by the interaction between the cognitive processing and the motor response. This is particularly evident when choice RT is used and compared to simple RT. Bradykinetic patients with PD have more specific impairment in choice RT, which involves a stimulus categorization and a response selection and reflects disturbance at more complex levels of cognitive processing. Ward and colleagues (1983b) found that of the various objective assessments of bradykinesia, the MT correlates best with the total clinical score, but it is not as sensitive an indicator of the overall motor deficit as the clinical rating.
Reduced dopaminergic function has been hypothesized to disrupt normal motor cortex activity, leading to bradykinesia. In recordings from single cortical neurons in free-moving rats, a decrease in firing rate correlated with haloperidol-induced bradykinesia, demonstrating that reduced dopamine action impairs the ability to generate movement and causes bradykinesia (Parr-Brownlie and Hyland, 2005). The premovement EEG potential (Bereitschaftspotential) is reduced in PD, probably reflecting inadequate basal ganglia activation of the supplementary motor area (Dick et al., 1989). On the basis of electromyographic (EMG) recordings in the antagonistic muscles of parkinsonian patients during a brief ballistic elbow flexion, Hallett and Khoshbin (1980) concluded that the most characteristic feature of bradykinesia was the inability to energize the appropriate muscles to provide a sufficient rate of force required for the initiation and maintenance of a large, fast (ballistic) movement. Therefore, PD patients need a series of multiple agonist bursts to accomplish a larger movement. Thus, the amount of EMG activity in PD is underscaled (Berardelli et al., 2001). Although many patients with PD complain of “weakness,” this subjective symptom is probably due to a large number of factors including bradykinesia, rigidity, fatigue, and also reduced power due to muscle weakness, particularly when lifting heavy objects (Allen et al., 2009).
Of the various parkinsonian signs, bradykinesia correlates best with a reduction in the striatal fluorodopa uptake measured by positron emission tomography (PET) scans and in turn with nigral damage (Vingerhoets et al., 1997). This is consistent with the finding that decreased density of SN neurons correlates with parkinsonism in the elderly, even without PD (Ross et al., 2004). PET scans in PD patients have demonstrated decreased 18F-fluorodeoxyglucose uptake in the striatum and accumbens-caudate complex roughly proportional to the degree of bradykinesia (Playford and Brooks, 1992). Studies performed initially in monkeys made parkinsonian with the toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Bergman et al., 1990) and later in patients with PD provide evidence that bradykinesia results from excessive activity in the subthalamic nucleus (STN) and the internal segment of globus pallidus (GPi) (Dostrovsky et al., 2002). Thus, there is both functional and biochemical evidence of increased activity in the outflow nuclei, particularly subthalamic nucleus and GPi, in patients with PD.
Tremor
By using the term shaking palsy, James Parkinson in his An Essay on the Shaking Palsy (1817) drew attention to tremor as a characteristic feature of PD. Indeed, some parkinsonologists regard rest tremor as the most typical sign of PD, and its absence should raise the possibility that the patient’s parkinsonism is caused by a disorder other than PD. The typical rest tremor has a frequency between 4 and 6 Hz, and the tremor is almost always most prominent in the distal part of an extremity. In the hand, the tremor has been called a “pill-rolling tremor.” In the head region, tremor occurs most commonly in the lips, chin and jaw, but while a common manifestation of essential tremor, head tremor is rare in PD (Roze et al., 2006; Gan et al., 2009). Some patients with PD complain of an internal, not visible, tremor, called “inner tremor.” Rest tremor of PD is often exacerbated during potential provocations, such as walking and counting backwards (Raethjen et al., 2008).
As pointed out below, presentation with tremor as the initial symptom often confers a favorable prognosis with slower progression of the disease and some have suggested the term “benign tremulous parkinsonism” for a subset of patients with minimal progression, frequent family history of tremor, and poor response to levodopa (Josephs et al., 2006; O’Suilleabhain et al., 2006). Rajput and colleagues (1991) noted that 100% of 30 patients with pathologically proven PD experienced some degree of rest tremor at some time during the course of their disease. However, in another clinical-pathologic study, only 76% of pathologically proven cases of PD had tremor (Hughes, et al., 1992b). In an expanded series of 100 pathologically proven cases of PD, tremor was present at onset in 69%; 75% had tremor during the course of the illness, and 9% lost their tremor late in the disease (Hughes et al., 1993).
Although rest tremor is a well-recognized cardinal feature of PD, many PD patients have a postural tremor that is more prominent and disabling than the classic rest tremor. Postural tremor without parkinsonian features and without any other known etiology is often diagnosed as essential tremor (ET), but isolated postural tremor may be the initial presentation of PD, and it may be found with higher-than-expected frequency in relatives of patients with PD (Brooks et al., 1992b; Jankovic et al., 1995; Jankovic, 2002; Louis et al., 2003). Jankovic and colleagues (1995) and others (Louis et al., 2003) have shown that relatives of patients with tremor-dominant PD have a significantly higher risk of having action tremor than relatives of patients with the PIGD form of PD, but it is not yet clear whether the isolated tremor in the relatives is ET or whether it represents an isolated manifestation of PD. The two forms of postural tremor, ET and PD, can be differentiated by a delay in the onset of tremor when arms assume an outstretched position. Most patients with PD tremor have a latency of a few seconds (up to a minute) before the tremor reemerges during postural holding, hence the term reemergent tremor (Jankovic et al., 1999b) (Video 4.1). In contrast, postural tremor of ET usually appears immediately after arms assume a horizontal posture. Since the reemergent tremor has a frequency similar to that of rest tremor and both tremors generally respond to dopaminergic drugs, reemergent tremor most likely represents a variant of the more typical rest tremor. In addition to the rest and postural tremors, a kinetic tremor, possibly related to enhanced physiologic tremor, may also impair normal reach-to-grasp movement (Wenzelburger et al., 2000).
While bradykinesia and rigidity are most likely associated with nigrostriatal dopaminergic deficit, the pathophysiology of PD rest tremor is probably more complicated and most likely results from dysfunction of both the striato-pallidal-thalamocortical and the cerebello-dentato-thalamocortical circuits (Boecker and Brooks, 2011). The pallidum, in particular, appears to play a fundamental role in generation of tremor as suggested by a 4-8 Hz GPi neuronal firing in primate models of parkinsonism, correlation of tremor severity with pallidal (but not striatal) dopamine depletion, and complete abolition or a marked improvement of tremor with GPi ablation or DBS (Helmich et al., 2011).
As a result of the abnormal neuronal activity at the level of the GPi, the muscle discharge in patients with PD changes from the normal high (40 Hz) to pulsatile (10 Hz) contractions. These muscle discharges, which may be viewed as another form of PD-associated tremor, can be auscultated with a stethoscope (Brown, 1997). 
Rigidity and flexed posture
Rigidity, tested by passively flexing, extending, and rotating the body part, is manifested by increased resistance throughout the range of movement. Cogwheeling is often encountered, particularly if there is associated tremor or an underlying, not yet visible, tremor. In 1926 Froment and Gardere published a series of papers based on their studies of parkinsonian rigidity, including the observation of enhanced resistance to passive movement of a limb about a joint detected during voluntary movement of a contralateral limb (“Froment’s maneuver”) (Broussolle et al., 2007). Rigidity may occur proximally (e.g., neck, shoulders, and hips) and distally (e.g., wrists and ankles). At times, it can cause discomfort and actual pain. Painful shoulder, possibly due to rigidity but frequently misdiagnosed as arthritis, bursitis, or rotator cuff, is one of the most frequent initial manifestations of PD (Riley et al., 1989; Stamey et al., 2008). In a prospective, longitudinal study of 6038 individuals, mean age 68.5 years, who participated in the Rotterdam study and had no dementia or parkinsonian signs at baseline, subjective complaints of stiffness, tremor, and imbalance were associated with increased risk of PD with hazard ratios of 2.11, 2.09, and 3.47, respectively (de Lau et al., 2006). During the mean 5.8 years of follow-up, 56 new cases of PD were identified.
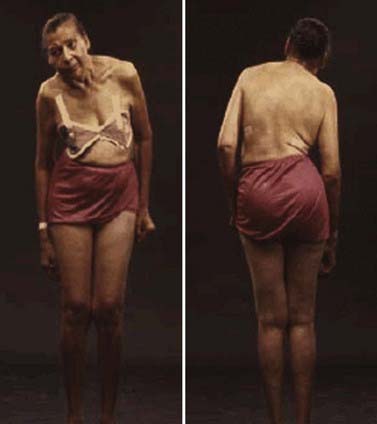
Rigidity is often associated with postural deformity resulting in flexed neck and trunk posture and flexed elbows and knees. But rigidity is a common sign in early PD, whereas flexed posture occurs later in the disease. Some patients develop “striatal hand” deformity, characterized by ulnar deviation of hands, flexion of the metacarpophalangeal joints, and extension of the interphalangeal joints (Fig. 4.3), and there may be extension of the big toe (“striatal toe”) or flexion of the other toes, which can be confused with arthritis (Jankovic and Tintner, 2001; Ashour et al., 2005; Ashour and Jankovic, 2006; Jankovic, 2007). Striatal toe was found to be present in 13 of 62 (21%) of patients with clinically diagnosed PD (Winkler et al., 2002).
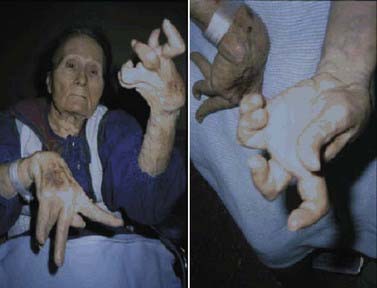
Other skeletal abnormalities include extreme neck flexion (“dropped head” or “bent spine”) (Oerlemans and de Visser, 1998; Askmark et al., 2001; Ashour and Jankovic, 2006; Kashihara et al., 2006; Gdynia et al., 2009; Oyama et al., 2009) and truncal flexion (camptocormia) (Djaldetti et al., 1999; Umapathi et al., 2002; Azher and Jankovic, 2005; Tiple et al., 2009; Sako et al., 2009). Askmark and colleagues (2001) found 7 patients out of 459 with parkinsonism who had a head drop attributed to neck extensor weakness. Myopathic changes on EMG were noted in all seven, and five patients who consented had abnormal muscle biopsy, with mitochondrial abnormalities in two. Isolated neck extensor myopathy was reported in other patients with anterocollis associated with parkinsonism (Lava and Factor, 2001; van de Warrenburg et al., 2007; Gdynia et al., 2009), although its true frequency in patients with PD, multiple system atrophy (MSA), and other parkinsonian disorders is unknown. The following etiologies have also been identified in various series of patients with head drop (head ptosis or anterocollis), bent spine, or camptocormia: dystonia, disproportionately increased tone in the anterior neck muscles resulting in fibrotic and myopathic changes, amyotrophic lateral sclerosis, focal myopathy, inclusion body myositis, polymyositis, nemaline myopathy, facioscapulohumeral dystrophy, myasthenia gravis, encephalitis, dopamine agonists (Uzawa et al., 2009), and valproate toxicity (Umapathi et al., 2002; Gourie-Devi et al., 2003; Schabitz et al., 2003; Azher and Jankovic, 2005; van de Warrenburg et al., 2007).
Camptocormia is characterized by extreme flexion of the thoracolumbar spine that increases during walking and resolves in supine position (Videos 4.2, 4.3, 4.4). It appears to be more common in patients with more severe PD and if they had prior vertebral surgery (Tiple et al., 2009). The term was coined during World War I when young soldiers who were apparently attempting to escape the stress of battle developed this peculiar posture, perhaps promoted by a stooped posture when walking in the trenches. There appear to be two possible mechanisms of camptocormia: (1) dystonia due to a central disorder and (2) extensor trunkal muscle myopathy (Bloch et al., 2006; Lepoutre et al., 2006; Melamed and Djaldetti, 2006; Gdynia et al., 2009). 
Other truncal deformities include scoliosis and tilting of the trunk, referred to as the Pisa syndrome (Villarejo et al., 2003) (Fig. 4.4). The axial dystonias resulting in scoliosis and camptocormia may improve with botulinum toxin injections into the paraspinal or rectus abdominis muscles (Azher and Jankovic, 2005; Bonanni et al., 2007). In some cases, dystonia may be the presenting symptom of PD, particularly the early-onset Parkinson disease (EOPD) variety such as is seen in patients with the parkin mutation (Lücking et al., 2000; Jankovic and Tintner, 2001; Hedrich et al., 2002). In addition, several autosomal recessive disorders, including PANK2, PLA2G6, ATP13A2, FBX07, TAF1, and PRKRA-associated neurodegeneration, are manifested by the dystonia–parkinsonism combination (Schneider et al., 2009). Another form of dystonia associated with PD is paroxysmal exercise-induced foot dystonia, which may be the presenting feature of young-onset PD (YOPD) (Bozi and Bhatia, 2003).
Loss of postural reflexes
Loss of postural reflexes is a characteristic feature in PD patients who exhibit the PIGD phenotype and usually occurs in more advanced stages of the disease along with freezing of gait and other symptoms that often lead to falling. One of the distinguishing features of PD fallers is their tendency to overestimate balance performance on functional reach testing compared to controls, and this overestimation worsens with worsening disease severity and when concurrently performing complex motor (e.g., carrying a tray) and cognitive tasks (e.g., performing mental arithmetic). In contrast to controls, PD patients are willing to sacrifice motor performance to complete competing tasks and make significantly more motor errors when performing a complex motor-cognitive task, whereas controls were more likely to preserve motor performance while sacrificing cognitive accuracy (Bloem et al., 2006). The loss of protective reactions further contributes to fall-related injuries. In one study, a fall in the past year, abnormal axial posture, cognitive impairment, and freezing of gait were independent risk factors for falls and predicted 38/51 fallers (75%) and 45/62 non-fallers (73%) (Latt et al., 2009). Additional measures contributing to falls include frontal impairment, poor leaning balance, and leg weakness. In another study, female gender, symmetrical onset, postural and autonomic instability appear to be the most reliable predictors of falls in PD (Williams et al., 2006). Using a battery of neurologic and functional tests in 101 patients with early PD and in an optimally medicated state, 48% reported a fall and 24% more than one fall in a prospective follow-up over 6 months (Kerr et al., 2010). The following measures provided the best sensitivity (78%) and specificity (84%) for predicting falls: UPDRS total score, total freezing of gait score, occurrence of symptomatic postural orthostasis, Tinetti total score, and extent of postural sway in the anterior-posterior direction.
The average period from onset of symptoms to the first fall in progressive supranuclear palsy (PSP) is 16.8 months, as compared to 108 months in PD, 42 months in MSA, 54 months in dementia with Lewy bodies, and 40.8 months in vascular parkinsonism. Many patients with postural instability, particularly when associated with flexed truncal posture (camptocormia), have festination, manifested by faster and faster walking as if chasing their center of gravity to prevent falling. When combined with axial rigidity and bradykinesia, loss of postural reflexes causes the patient to collapse into the chair when attempting to sit down. The pull test (pulling the patient by the shoulders) is commonly used to determine the patient’s degree of retropulsion or propulsion (see above) (Visser et al., 2003; Hunt and Sethi, 2006; Valkovic et al., 2008). Alterations in cholinergic rather than dopaminergic neurotransmission have been implicated in disturbed balance and falls associated with PD, partly because of evidence that gait control depends on cholinergic system-mediated higher-level cortical and subcortical processing, including pedunculopontine nucleus (PPN) function. This is supported by a cross-sectional study of 44 patients with PD without dementia and 15 control subjects who underwent a clinical assessment and [11C]methyl-4-piperidinyl propionate (PMP) acetylcholinesterase (AChE) and [11C]dihydrotetrabenazine (DTBZ) vesicular monoamine transporter type 2 (VMAT2) brain PET imaging (Bohnen et al., 2009). The study found reduced cortical AChE hydrolysis rates demonstrated in the PD fallers (−12.3%) compared to PD nonfallers (−6.6%) and control subjects (P = 0.0004). In another study, involving 22 normal controls, 12 patients with PD, 13 with MSA-P, and 4 with PSP, PET with [11C]PMP showed a significant decrease in AChE activity in most cerebral cortical regions in PD and MSA-P, and a nonsignificant decrease in PSP. On the other hand, subcortical cholinergic activity was significantly more decreased in MSA-P and PSP than in PD. The authors suggested that the more substantial decrease in subcortical AChE in MSA-P and PSP reflects greater impairment in the pontine cholinergic group (PPN) and may account for the greater gait disturbances in the early stages of these two disorders compared to PD (Gilman et al., 2010). Thus cholinergic hypofunction, possibly related to PPN degeneration, may be contributing to falls in patients with PD (Thevathasan and Aziz, 2010).
Freezing
One of the most disabling symptoms of PD is freezing, also referred as motor blocks, considered by some as a form of akinesia (loss of movement) (Giladi et al., 1997, 2001; Giladi and Nieuwboer, 2008; Morris et al., 2008) (Videos 4.5, 4.6). Although it most often affects the legs when walking, it can also involve upper limbs and the eyelids (apraxia of eyelid opening or eyelid closure) (Boghen, 1997). Freezing consists of sudden, transient (a few seconds) inability to move. It typically causes start hesitation when initiating walking and the sudden inability to move feet (as if glued to the ground) when turning or walking through narrow passages (such as the door or the elevator) (Almeida and Lebold, 2010), when crossing streets with heavy traffic, or when approaching a destination (target hesitation). Freezing is the most common cause of falls in patients with PD that can result in injuries, including hip fractures. Patients often adopt a variety of cues or tricks to overcome the freezing attacks: marching to command (“left, right, left, right”), stepping over objects (the end of a walking stick, a pavement stone, cracks in the floor, etc.), walking to music or a metronome, shifting body weight, rocking movements, and others (Dietz et al., 1990; Fahn, 1995; Marchese et al., 2001; Rubinstein et al., 2002; Suteerawattananon et al., 2004; Nieuwboer, 2008). “Off” gait freezing was found to correlate with dopa-responsive abnormal discriminatory processing as determined by abnormally increased temporal discrimination threshold (Lee et al., 2005). Freezing may be a manifestation of the “off” phenomenon in PD patients who fluctuate but may also occur during “on” time (“on freezing”), independent of bradykinesia and tremor (Bartels et al., 2003). Based on responses by 6620 patients to a questionnaire sent to 12 000 members of the German Parkinson Association, 47% of patients reported freezing, and it was present more frequently in men than women and less frequently in patients who considered tremor as their main symptom (Macht et al., 2007). When freezing occurs early in the course of the disease or is the predominant symptom, a diagnosis other than PD should be considered. Disorders associated with prominent freezing include progressive supranuclear palsy (PSP), MSA, and vascular (lower body) parkinsonism (FitzGerald and Jankovic, 1989; Elble et al., 1996; Winikates and Jankovic, 1999; Jankovic et al., 2001). Freezing has been thought to be related to noradrenergic deficiency as a result of degeneration of the locus coeruleus (Zarow et al., 2003), as suggested by possible response to noradrenergic agents such as L-threo-dihydroxy-phenylserine, or DOPS (Narabayashi, 1999). Neurophysiologic studies in monkeys treated with MPTP found that dopamine depletion is associated with impaired selection of proprioceptive inputs in the supplementary motor area, which could interfere with motor planning and may be related to motor freezing (Escola et al., 2002). Integrating EMG signals over real time while recording EMG activity from lower extremities before and during freezing, Nieuwboer and colleagues (2004) showed significantly abnormal timing in the tibialis anterior and gastrocnemius muscles, although reciprocity is preserved. Thus, before freezing, the tibialis anterior and gastrocnemius contract prematurely, and the duration of contraction is shortened in the tibialis anterior, but the amplitude of the EMG burst is increased (probably a compensatory strategy pulling the leg into swing), whereas the contraction is prolonged in the gastrocnemius during the actual swing phase. Isolated freezing usually suggests a diagnosis other than PD and may be present in atypical forms of parkinsonism or brainstem strokes (Kuo et al., 2008). The pathologic involvement of brainstem in patients with pure akinesia and gait freezing is suggested by decreased glucose metabolism on PET scans in the midbrain of such patients, similar to the findings in patients with PSP (Park et al., 2009). Neither medical (Giladi, 2008) nor surgical (Ferraye et al., 2008; Nashatizadeh and Jankovic, 2008) treatments generally provide satisfactory control of freezing. 
Other motor abnormalities
Some patients exhibit the reemergence of primitive reflexes attributed to a breakdown of the frontal lobe inhibitory mechanisms that are normally present in infancy and early childhood, hence the term release signs (Vreeling et al., 1993; Thomas, 1994; Rao et al., 2003). The glabellar tap reflex, also known as Meyerson sign, has often been associated with PD. However, its diagnostic accuracy has not been subjected to rigorous studies. The glabellar tap reflex is elicited through repeated stimuli to the glabellar region of the forehead, inducing concomitant blinking with each tap. In the normal subject, the reflex blinking habituates or the subject stops blinking with each stimulus tap after the second to fifth tap. Brodsky and collegues (2004) examined the glabellar reflex and the palmomental reflex in 100 subjects, which included patients with PD (n = 41), patients with PSP (n = 12), patients with MSA (n = 7), and healthy, age-matched controls (n = 40). Using a standardized protocol and a “blinded” review of videotapes, we found that (1) both reflexes were present significantly more frequently in patients with PD as compared to normal controls; (2) glabellar, but not palmomental, reflex was more frequently present in patients with PSP than in controls; (3) there was no difference in the frequency of these reflexes between normal controls and patients with MSA; (4) the two reflexes occurred with similar frequency among the three parkinsonian disorders; (5) glabellar, but not palmomental, reflex correlated with parkinsonian motor deficit; and (6) the primitive reflexes correlated with mental deficit. While relatively sensitive signs of parkinsonian disorders, particularly PD, these primitive reflexes lack specificity, as they do not differentiate among the three most common parkinsonian disorders (Brodsky et al., 2004). Abnormal spontaneous blinking, particularly the longer pauses between closing and opening phase, compared to normal controls suggests that the decreased blinking in PD reflects underlying bradykinesia (Agostino et al., 2008). In addition to these primitive reflexes, there are other “frontal” and “cortical disinhibition” signs, such as the applause sign (Wu et al., 2008), but none of them are specific for PD.
Besides the classic cardinal signs, there are many other motor abnormalities that may be equally or even more disabling. One of the most prominent features of motor impairment in PD is the inability to perform multiple tasks simultaneously. Using functional magnetic resonance imaging (fMRI), Wu and Hallett (2008) found that during dual task execution, greater activity was recorded in the precuneus region, cerebellum, premotor area, and parietal and prefrontal cortex. They concluded that difficulties in dual task performance in PD were associated with limited attentiveness and defective central executive function, and that training may improve the performance. The bulbar symptoms (dysarthria, hypophonia, dysphagia, and sialorrhea) are thought to result from orofacial-laryngeal bradykinesia and rigidity (Hunker et al., 1982). PD-associated speech and voice impairment, often referred to as hypokinetic dysarthria, is characterized by low volume (hypophonia), uniform (monotonous) loudness and pitch (aprosody), imprecise consonants, hesitation, and short rushes of speech (tachyphemia). Other speech characteristics include a variable (abnormally slow or increased) speech rate, palilalia, and stuttering. PD patients have been found to have higher speech acceleration than controls and a significant reduction in the number of pauses, indicating abnormal speech rate and rhythm (Skodda and Schlegel, 2008). A history of childhood stuttering that had remitted can subsequently recur with onset of PD, suggesting an involvement of the dopaminergic system in this speech disorder (Shahed and Jankovic, 2001). When speech therapy designed to stimulate increased vocal fold adduction with instructions to “think loud, think shout,” the Lee Silverman Voice Treatment (Ramig et al., 2001), was compared with “speak loud and low,” the Pitch Limiting Voice Treatment (de Swart et al., 2003), the two methods produced the same increase in loudness, but the latter method was found to prevent strained voicing. Other treatment strategies for PD-related dysarthria include the use of various verbal cues to regulate speech volume (Ho et al., 1999), but deep brain stimulation has a variable effect (Pinto et al., 2004). The low-volume voice in PD has been attributed in part to vocal fold bowing due to loss of muscle mass and control (Schulz et al., 1999), and augmentation of vocal folds with collagen injections provides improvement in voice quality and has a significantly beneficial impact on quality of life (Hill et al., 2003). Respiratory difficulties result from a variety of mechanisms, including a restrictive component due to rigid respiratory muscles and levodopa-induced respiratory dyskinesias (Rice et al., 2002).
In addition to categorization of patients into clinical subtypes, there is a growing appreciation for differences in clinical presentation depending on genetic background. Thus patients with parkin mutations (PARK2), who account for nearly a third of patients with early-onset PD, tend to develop levodopa-induced dyskinesias and hallucinations relatively early in the course of the disease. They also may present with dystonic gait, cervical dystonia, dopa-responsive dystonia, hemiparkinsonism–hemiatrophy, freezing, festination, retropulsion, leg tremor at rest and on standing, marked sleep benefit, hyperreflexia, ataxia, peripheral neuropathy, and dysautonomia (Klein and Lohmann, 2009). Carriers of LRRK2 G2019S mutation are more likely to manifest the PIGD subtype of PD rather than the tremor-dominant phenotype, although in contrast to the PIGD in patients with sporadic PD, LRRK2 patients tend to have a much slower, less aggressive course (Alcalay et al., 2009; Dächsel and Farrer, 2010). Other studies have confirmed that in comparison with genetically undefined patients, LRRK2 mutation carriers had more severe motor symptoms, a higher rate of dyskinesia, and less postural tremor, whereas PINK1 mutation carriers have younger age at onset and slower progression, but similar to LRRK2 PD patients have an increased rate of drug-induced dyskinesia and a lower rate of postural tremor (Nishioka et al., 2010).
Nonmotor manifestations
Although James Parkinson in his original description focused on the motor symptoms, he also drew attention to several nonmotor features, including problems associated with sleep and gastrointestinal function (Parkinson, 1817). Traditionally viewed as primarily a motor disorder, there is growing recognition that nonmotor symptoms of PD, which occur in 88% of all patients, are as troublesome if not more so than the classic motor features (Simuni and Sethi, 2008). The nonmotor manifestations and fluctuations in nonmotor symptoms have been found to be more disabling than the motor symptoms in 28% of PD patients (Witjas et al., 2002). These nonmotor, nondopaminergic symptoms have been largely ignored, but several recent studies have highlighted their frequency and their serious impact on quality of life, particularly in more advanced stages of the disease (Lang and Obeso, 2004; Chaudhuri et al., 2006a, 2006b, 2007; Ahlskog, 2007; Martinez-Martin et al., 2007; Pfeiffer, 2007; Lim et al., 2009). The Sydney Multicenter Study showed that PD patients treated with “modern” initial therapy continue to die at a rate in excess of their peers, with only one-third of original study subjects remaining alive at 15 years after diagnosis, and most were disabled more by their nonmotor than motor symptoms: 84% experienced cognitive decline with 48% meeting diagnostic criteria for dementia, 58% were unable to live alone, and 40% were in long-term care facilities (Hely et al., 2005). After 20 years’ follow-up, only 36 (26%) survived and the standardized mortality ratio reached 3.1 (Hely et al., 2008). Of the 30 included in this longitudinal study, 100% had levodopa-induced dyskinesia and end of dose failure, dementia was present in 83%, and 48% were in nursing homes. Other problems included excessive daytime sleepiness in 70%, falls in 87%, freezing in 81%, fractures in 35%, symptomatic postural hypotension in 48%, urinary incontinence in 71%, moderate dysarthria in 81%, choking in 48%, and hallucinations in 74%. Of the 87 patients followed prospectively in the Sydney study whose brain was examined at autopsy, the final diagnosis was PD in 29, PD with dementia in 52, and dementia with Lewy bodies (DLB) in 6 (Halliday et al., 2008). The clinical-pathologic correlations suggested that there were three groups of patients: (1) younger-onset patients with a typical PD clinical course; brainstem Lewy bodies predominate in those surviving to 5 years, and by 13 years, 50% of cases have a limbic distribution of Lewy bodies; (2) older-onset PD cases with shorter survival and with higher Lewy body loads and additional plaque pathology; and (3) early malignant, dementia-dominant syndrome and severe neocortical disease, consistent with DLB. In a multicenter study of 1072 consecutive patients with PD in 55 Italian centers, the so-called Priamo study, 98.6% of patients reported a mean of 7.8 (range 0–32) nonmotor symptoms, such as fatigue (58%), anxiety (56%), leg pain (38%), insomnia (37%), urinary urgency and nocturia (35%), drooling of saliva (31%) and difficulties in maintaining concentration (31%) (Barone et al., 2009). Apathy was the symptom associated with worse PDQ-39 score but presence of fatigue, attention/memory, and psychiatric symptoms also had a negative impact on quality of life. The nonmotor features associated with PD are presumably related to involvement of the nondopaminergic systems and even pathology outside the central nervous system (Djaldetti et al., 2009).
Autonomic dysfunction
Autonomic failure is typically associated with MSA and may be the presenting feature of that disease, but it may also herald the onset of PD (Kaufmann et al., 2004; Mostile and Jankovic, 2009). In contrast to PD associated with dysautonomia, due to predominantly peripheral (ganglionic and postganglionic) involvement, in MSA the primary lesion is preganglionic; also dysautonomic symptoms are more severe at baseline and become more global in MSA as compared to PD (Lipp et al., 2009). Rating scales for dysautonomia associated with PD have been developed (Evatt et al., 2009). Dysautonomia, such as orthostatic hypotension, sweating dysfunction, sphincter dysfunction, and sexual impotence occur frequently in patients with PD (Senard et al., 1997; Swinn et al., 2003). In one study, 7 of 51 (14%) patients with early, untreated PD had a decrease of more than 20 mmHg in systolic blood pressure (Bonuccelli et al., 2003). Another community-based study of a cohort of PD patients showed that 42 of 89 (47%) met the diagnostic criteria for orthostatic hypotension (Allcock et al., 2004). Orthostatic hypotension, however, is not often detected in the clinic. Although the symptom of orthostatic lightheadedness has a relatively high specificity, it seems to have low sensitivity in predicting orthostatic hypotension, partly because it is more likely to occur after tilting than on standing, and is often delayed by longer than the recommended 3 minutes (Jamnadas-Khoda et al., 2009). Autonomic symptoms, particularly orthostatic hypotension, seem to be more common in the PIGD form of PD (Allcock and et al., 2006). Autonomic symptom severity was associated with more motor dysfunction, depressive symptoms, cognitive dysfunction, psychiatric complications, nighttime sleep disturbances, and excessive daytime sleepiness (P < 0.01) (Verbaan et al., 2007a). While dysautonomia is typically associated with MSA, it may also be prominent in PD, although autonomic testing might not always differentiate between PD and MSA (Riley and Chelimsky, 2003).
Orthostatic hypotension in patients with PD has been traditionally attributed to dopaminergic therapy, but recent studies have provided evidence that orthostatic hypotension in PD is due to failure of reflexive sympathetically mediated cardiovascular stimulation from sympathetic denervation, as demonstrated by markedly decreased 6-[18F]-fluorodopamine-derived radioactivity in septal and ventricular myocardium (Goldstein et al., 2002). This sympathetic nervous system deficit involved postganglionic catecholaminergic, not cholinergic, nerves (Sharabi et al., 2003).
Sweating dysfunction, hyperhidrosis, and to a lesser extent hypohidrosis, were reported by 64% of patients with PD as compared to 12.5% of controls (P < 0.005) (Swinn et al., 2003). These symptoms did not correlate with the severity of the disease but occurred most frequently during the “off” periods and during “on with dyskinesia” periods. Because sudomotor skin response was reduced in the palms, the axial hyperhidrosis has been suggested to be a compensatory phenomenon for reduced sympathetic function in the extremities (Schestatsky et al., 2006). Sweating may be a particularly troublesome symptom during wearing off (Pursiainen et al., 2007). The presence of α-synuclein deposits in the dermis of a patient with pure autonomic failure provides evidence that this disorder as well as other disorders associated with autonomic failure (e.g., PD, DLB, and MSA) should be viewed as variant synucleinopathies (Kaufmann and Goldstein, 2010; Shishido et al., 2010).
Bladder and other urologic symptoms are frequent in PD and are among the most common complaints requiring medical attention (Blackett et al., 2009; Sakakibara et al., 2010). One survey found that over one-fourth of men with PD had urinary difficulty, most often causing urinary urgency (Araki and Kuno, 2000). In one study, urge episodes and urge incontinence were observed in 53% and 27% of the patients with PD, respectively, and detrusor overactivity in 46% of the patients with PD, which was less prevalent than in patients with dementia with Lewy bodies and Alzheimer disease, while mean voided volume, free flow, cystometric bladder capacity, and detrusor pressor were similar in the groups (Ransmayr et al., 2008).
Despite the possibility of hypersexuality, usually related to dopaminergic drugs, many patients with PD have sexual dysfunction (Celikel et al., 2008; Meco et al., 2008; Hand et al., 2010). In a review of sexual functioning of 32 women and 43 men with PD, women reported difficulties with arousal (87.5%), reaching orgasm (75.0%), and sexual dissatisfaction (37.5%) (Bronner et al., 2004). Men reported erectile dysfunction (68.4%), sexual dissatisfaction (65.1%), premature ejaculation (40.6%), and difficulties reaching orgasm (39.5%). Reduced sexual drive and dissatisfaction with orgasm was particularly common in female PD patients (Celikel et al., 2008). Among 90 patients with PD, loss of libido was reported by 65.6%, and 42.6% of men also complained of erectile dysfunction (Kummer et al., 2009). Aging, female gender, lower education, and depression were significantly associated with decreased sexual desire. Premorbid sexual dysfunction may contribute to cessation of sexual activity during the course of the disease (among 23.3% of men and 21.9% of women). Associated illnesses, use of medications, motor difficulties, depression, anxiety, and advanced stage of PD contributed to sexual dysfunction.
Drooling (sialorrhea) is one of the most embarrassing symptoms of PD (Chou et al., 2007). While some studies have shown that PD patients actually have less saliva production (Proulx et al., 2005) than normal controls, others have suggested that the excessive drooling is due to a difficulty with swallowing (Bagheri et al., 1999). Salivary sympathetic denervation, however, could not be demonstrated by 6-[18F]-fluorodopamine scanning (Goldstein et al., 2002). Dysphagia (Hunter et al., 1997) along with delayed gastric emptying (Hardoff et al., 2001) and constipation (Ashraf et al., 1997; Bassotti et al., 2000; Winge et al., 2003; Cersosimo and Benarroch, 2008) represent the most frequent gastrointestinal manifestations of PD. In addition to constipation, PD patients often experience pharyngeal and esophageal dysphagia and 60% have evidence of delayed gastric emptying (Krygowska-Wajs et al., 2009).
When PD patients were compared to healthy controls, those with PD were found to swallow significantly more often during inhalation, at low tidal volumes, and exhibited significantly more post-swallow inhalation (Gross et al., 2008). Impaired coordination of breathing and swallowing may contribute to the high frequency of aspiration pneumonia in PD.
Gastrointestinal dysfunction in PD has been attributed to many mechanisms such as involvement, including neurodegeneration and the presence of Lewy bodies, of the dorsal motor nucleus of the vagus, paravertebral sympathetic ganglia, and intrinsic neurons of the enteric nervous system (Cersosimo and Benarroch, 2008). On the basis of information on the frequency of bowel movements in 6790 men in the Honolulu Heart Program, Abbott and colleagues (2001) concluded that infrequent bowel movements are associated with increased risk for future PD. Based on the observation from the Honolulu-Asia Aging Study that bowel frequency was lower in subjects who were found to have incidental Lewy bodies in their brains at postmortem examination than in controls, the investigators suggested that constipation was one of the earliest symptoms of PD (Abbott et al., 2007). Also, constipation was associated with low SN neuron density (Petrovitch et al., 2009). Based on a review of Mayo Clinic medical records of 196 case-control pairs (N = 392), constipation preceding PD was more common in cases than in controls (odds ratio 2.48; P = 0.0005) (Savica et al., 2009). Constipation in patients with PD is associated with slow colonic transit, weak abdominal strain, decreased phasic rectal contraction, and paradoxical sphincter contraction on defecation (Sakakibara et al., 2003). Dermatologic changes such as seborrhea, hair loss, and leg edema may represent evidence of peripheral involvement in PD, although some of these changes may be exacerbated by anti-PD drugs (Tabamo and Di Rocco, 2002; Tan and Ondo, 2000).
Autonomic complications, coupled with motor and mental decline, contribute to a higher risk of hospitalization and nursing home placement. Examination of hospital records of 15 304 cases of parkinsonism and 30 608 age- and sex-matched controls showed that PD patients are six times more likely to be admitted to hospital with aspiration pneumonia than are nonparkinsonian controls (Guttman et al., 2004). Other comorbid medical conditions significantly more common in patients with PD include fractures of the femur, urinary tract disorders, septicemia, and fluid/electrolyte disorders. But similarly to other reports (Jansson and Jankovic, 1985; Gorell et al., 1994; Vanacore et al., 1999; Inzelberg and Jankovic, 2007), this study showed that cancer might be less common in patients with PD, with the major exception being malignant melanoma with an almost twofold increased risk (Olsen et al., 2005) and a higher risk of family history of melanoma (Gao et al., 2009).
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


