Pharmacokinetics
Emilio Perucca
Introduction
Pharmacokinetics is the study of changes in the concentration of drugs or drug metabolites within the body as a function of time. Its clinical importance stems from the fact that the concentration of the active principle at the site of action is generally the primary determinant of therapeutic and toxic effects. Therefore, pharmacokinetic principles play a critical role in determining the time of onset, magnitude, and duration of drug action.
For some drugs (e.g., carbamazepine), there seems to be a direct relationship between drug concentration in blood and the intensity of pharmacologic effect.84 In other cases, the relationship between kinetics and effects may be more complex. The duration of action of vigabatrin, for example, may outlast significantly the presence of the drug in the body owing to the fact that the effect is mediated by irreversible inhibition of the enzyme γ-aminobutyric acid (GABA) transaminase.23 Although in such cases a direct relationship between plasma (or brain) concentration and pharmacologic effect is not anticipated, it is still true that the effects ultimately depend on an adequate amount of drug reaching its site of action in the brain.
Drug kinetics result from a complex interplay among three basic processes: (a) absorption, (b) distribution, and (c) elimination. Characterization of these processes has proved to be invaluable for the rational use of antiepileptic drugs. Therapeutic advances related to an understanding of kinetics include recognition of the need to individualize dose to compensate for interpatient differences in drug disposition, selection of rational dosing schedules and dosing intervals, development of innovative drug formulations, identification of clinically relevant pharmacokinetic interactions, and use of plasma drug level monitoring as a guide to adjustments in dose.17,69,83
Absorption
The term absorption is used to describe the processes involved in the transfer of the drug from the site of administration to the bloodstream. The most important factors affecting absorption are the route of administration, the physical-chemical properties of the drug, the formulation used, and the pathophysiologic conditions of the patient.
The term bioavailability has been introduced to define how much of a drug reaches the systemic circulation after extravascular administration and the rate at which this takes place. Bioavailability primarily depends on the rate and extent of absorption, but it is also influenced by biotransformation or degradation, which may occur before the drug reaches the systemic circulation. For example, a compound may be fully absorbed from the gastrointestinal tract and yet be incompletely bioavailable if a significant fraction of the dose is metabolized during its first passage through the liver (the so-called first-pass effect).
Antiepileptic drugs are normally administered orally, but in particular situations the intravenous, intramuscular, and rectal routes are used alternatively. Other routes are used far less commonly; for midazolam, in particular, a number of studies have suggested that intranasal47 or buccal38 administration may result in rapid achievement of serum drug levels sufficient for anticonvulsant activity.
Gastrointestinal Absorption
Several processes must take place before an orally administered drug reaches the systemic circulation. These include (a) disintegration of the pharmaceutical preparation or release from the formulation (in the case of solid dose forms), (b) dissolution into the gastrointestinal fluids, (c) transfer across the gastrointestinal mucosa to the local bloodstream (a process that for some drugs may be affected by the activity of transporter systems located on the intestinal epithelium), and (d) possible presystemic degradation (or biotransformation) in the gastrointestinal tract or in the liver.66
Although some drugs can be partially absorbed from the stomach (especially acidic compounds, which are nonionized and lipophilic at low pH values), absorption takes place mostly in the gut and therefore may be slowed when gastric emptying is delayed. In general, absorption is faster after ingestion of syrups or solutions than after intake of solid forms. Formulations produced by different manufacturers may differ in rate or extent of absorption, resulting in potentially important differences in bioavailability. For example, intoxication or loss of seizure control has occurred following the substitution of one formulation for another that was subsequently found to have a different bioavailability. Such problems have been reported with many antiepileptic drugs, including phenytoin,56 carbamazepine,21 and primidone.103 Regulations for the licensing of new formulations, including generics, recently have been tightened in several countries. Therefore, problems resulting from bioinequivalence are less likely to occur when a brand product is substituted with a generic product intended to ensure a comparable absorption profile.82
At times, formulations are especially designed to modify advantageously the release of the active principle into gastrointestinal contents. For example, drugs such as carbamazepine and valproic acid, which are absorbed and eliminated rapidly, are also available as sustained-release dose forms; these may be useful for attenuating excessive fluctuations in plasma drug levels and allowing less frequent dosing.9 Sodium valproate may also be prescribed in the form of enteric-coated tablets, which are designed to avoid disintegration in the stomach and so prevent gastric irritation. Enteric-coated tablets do not behave as sustained-release forms because after dissolution of the coating, absorption proceeds very rapidly. Enteric coating, however, does modify absorption characteristics significantly. In fact, although small particles (<2 mm in diameter) can pass through the pylorus irrespective of the degree of
gastric filling, the motility pattern of fasting is needed for larger particles to leave the stomach.92 Ingestion of enteric-coated preparations after a meal may result in a long delay (sometimes up to 10 hours or even longer) before the tablet leaves the stomach and reaches the duodenum, where the coating dissolves and absorption occurs rapidly. This phenomenon explains the long latency that may occur before valproic acid is absorbed from enteric-coated tablets taken with or after a meal (Fig. 1).42
gastric filling, the motility pattern of fasting is needed for larger particles to leave the stomach.92 Ingestion of enteric-coated preparations after a meal may result in a long delay (sometimes up to 10 hours or even longer) before the tablet leaves the stomach and reaches the duodenum, where the coating dissolves and absorption occurs rapidly. This phenomenon explains the long latency that may occur before valproic acid is absorbed from enteric-coated tablets taken with or after a meal (Fig. 1).42
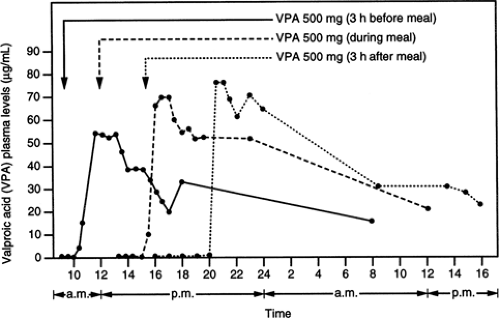 FIGURE 1. Effect of a meal on the absorption of valproic acid (VPA) from enteric-coated tablets in a representative individual. The same dose (one 500-mg tablet) was taken before a meal, during a meal, and 3 hours after a meal. Arrows indicate the time of drug intake. Note the long latency between drug intake and initiation of absorption when the drug was taken during or after a meal. (From Levy RH, Cenraud B, Loiseau P, et al. Meal-dependent absorption of enteric-coated sodium valproate. Epilepsia. 1980;21:273–280, with permission.) |
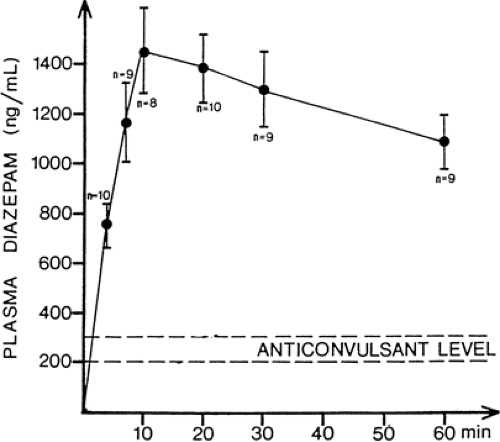 FIGURE 2. Rapid absorption of diazepam from a commercial solution (0.7 mg/μg) after rectal instillation in infants. Anticonvulsant activity is considered to develop when plasma concentration reaches the range of 200 to 300 ng/mL. (Modified from Knudsen FU. Plasma diazepam in infants after rectal administration in solution and by suppository. Acta Paediatr Scand. 1977;66:563–567.) |
Irrespective of the formulation used, concurrent intake of food may have a profound influence on drug absorption. For some drugs (e.g., azithromycin, albendazole, griseofulvin), the rate and extent of absorption can be enhanced by food intake, whereas for others (e.g., clodronate, etidronate, tetracycline), bioavailability may be reduced drastically when they are taken after a meal.87 Absorption may also be affected by drug interactions in the gastrointestinal tract, including interactions affecting the activity of transporter systems such as P-glycoprotein (see Chapter 110).
Rectal Absorption
The rectal route may provide a useful alternative when oral administration is not feasible because of, for example, palat-ability problems, nausea and vomiting, or a patient’s inability to cooperate during ongoing seizures. For drugs that show a prominent first-pass effect in the liver, rectal administration may enhance bioavailability because only the superior hemorrhoidal vein drains into the portal system.
The efficiency of rectal absorption is influenced primarily by the physical-chemical properties of the drug (lipophilic compounds are usually well absorbed) and by the type of pharmaceutical formulation.96 Suppositories may be associated with lower rates and extent of absorption, whereas absorption from rectal solutions or rectal capsules can be quite efficient. In the case of diazepam, anticonvulsant concentrations can be achieved within minutes of rectal instillation of the commercial solution (Fig. 2). The rapid absorption of rectally administered diazepam can be exploited when, for example, prolonged
seizures must be managed by nonmedical personnel or intermittent prophylaxis is needed against the recurrence of febrile convulsions.36
seizures must be managed by nonmedical personnel or intermittent prophylaxis is needed against the recurrence of febrile convulsions.36
Intramuscular Absorption
The rate of absorption following intramuscular injection is determined by the solubility properties of the drug, the pharmaceutical characteristics of the formulation, and the degree of local blood perfusion. The latter, in turn, varies with site of injection (blood flow and absorption rate are greater in the deltoid region than in the buttock), hemodynamic state (absorption may be inadequate in hypotensive patients), and injection technique (massaging the muscle after the injection, e.g., may hasten absorption).
With some drugs, such as diazepam, intramuscular absorption may be slow and erratic.32 For this reason, diazepam should not be given intramuscularly for the treatment of status epilepticus. Midazolam, a hydrophilic benzodiazepine, is absorbed from the muscle at a much faster rate than diazepam, and intramuscular midazolam is a viable option for the treatment of acute seizures.80
The slow and prolonged absorption of some drugs from muscle should be taken into account when the route of drug administration is temporarily switched from oral to intramuscular to provide therapeutic cover when a patient is transiently unable to take medications orally. For example, if phenytoin needs to be given intramuscularly for a few days, a dose 50% higher than the oral maintenance dose may be needed to compensate for inadequate intramuscular absorption. On resumption of oral therapy, half of the original oral dose should then be given for a period of time equal to the period of intramuscular administration so that toxicity resulting from delayed absorption from muscle stores is prevented.99 Whenever possible, however, intramuscular administration of phenytoin should be avoided because it may cause local pain and muscle damage. If the patient cannot take phenytoin orally, the intravenous route is a preferable alternative. Another alternative is to switch temporarily to fosphenytoin, which, unlike phenyt-oin, is efficiently absorbed after intramuscular injection (see Chapter 154).
Distribution
After reaching the circulation, a drug undergoes distribution within blood components and body tissues. The rate and extent of penetration into individual tissues depend on a number of factors, such as the physical-chemical properties of the drug, the degree of drug binding to plasma and tissue components, the degree of tissue perfusion (blood flow), the presence of biologic barriers to tissue penetration (e.g., the blood–brain barrier), and the activity of transporter systems that may limit drug access to certain tissues and/or certain cellular or subcellular compartments. Compared with highly water-soluble drugs, lipophilic compounds generally exhibit more extensive tissue penetration, particularly in organs with a high lipid content, such as the brain. Distribution primarily occurs by passive diffusion, although in some cases drug transfer may be regulated by active transport mechanisms.
Table 1 Plasma protein binding of major antiepileptic drugs | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
One estimate of the extent of drug penetration into tissues is provided by the volume of distribution. This parameter, which is usually normalized for body weight, can be defined as the virtual volume in which the total amount of drug present in the organism after completion of distribution processes needs to be dissolved to yield a concentration identical to that found in plasma. A volume of distribution of about 1 L/kg indicates that the drug is found in tissues at an average concentration equivalent to that in plasma. A volume of distribution <1 L/kg indicates that the tissue concentration is on average lower than the plasma concentration. Conversely, a volume of distribution in >1 L/kg indicates a high tissue concentration relative to the concentration in plasma. Most antiepileptic drugs have volumes of distribution between 0.6 and 1.2 L/kg, indicating that on average their tissue concentration is 60% to 120% of their plasma concentration.67,68 There are exceptions to this rule, however, the most notable example being valproic acid; the small volume of distribution of this drug (about 0.15 L/kg) indicates its relatively poor tissue penetration.73 An important concept is that the volume of distribution does not provide any information about drug concentration at specific sites; in fact, any given average tissue concentration may result from drug accumulation at extremely high levels in some organs, with other tissues receiving little or no drug exposure. More specifically, the volume of distribution does not provide any indication about drug concentration in the brain: Some drugs have a high volume of distribution and yet fail to reach the central nervous system because of inability to cross the blood–brain barrier.
Binding to Plasma Proteins
Many drugs are significantly bound to plasma proteins (Table 1). Although the binding is generally of a loose and reversible type, the drug–protein complex is too large to move freely across biologic membranes, and therefore it is only the unbound moiety that creates the diffusion gradient driving drug molecules from the blood into the tissues. Albumin is the most important protein available for the transport of acidic drugs, such as phenytoin and valproic acid, whereas α1-acid glycoprotein is primarily involved in the transport of basic compounds, such as chlorpromazine and lidocaine. Some drugs (e.g., carbamazepine), bind to a significant extent to both albumin and α1-acid glycoprotein.2
Binding to plasma proteins may have a significant influence on drug kinetics. First, the protein-bound drug represents a reservoir within the bloodstream that tends to increase the concentration in plasma relative to the concentration in tissues. Second, drug binding to proteins may affect the rate of drug elimination. In the case of low-clearance drugs, only the unbound drug can be cleared, and therefore protein binding tends to restrict the efficiency of the elimination process. Conversely, in the case of high-clearance drugs, both the unbound and the protein-bound drug can be cleared; under these circumstances, protein binding enhances elimination by increasing the amount of drug delivered to the eliminating organ.77,101
The binding of drugs to plasma proteins may vary considerably among patients because of differences in protein concentrations or the presence of endogenous and exogenous substances competing at protein-binding sites. Because only the unbound drug is in equilibrium with drug molecules at receptor sites, pharmacologic effects usually correlate better with concentration of unbound drug than with total plasma concentration. Therefore, any condition leading to a change in unbound fraction will alter the relationship between plasma drug concentration and therapeutic effect, and this needs to be taken into account when drug levels are interpreted in clinical practice.74 For example, a reduced binding of phenytoin to plasma proteins is observed in neonates; the elderly; women in late pregnancy; patients with other hypoalbuminemic conditions, hyperbilirubinemia, or uremia; and patients undergoing concomitant treatment with displacing drugs, such as valproic acid.63 Under these circumstances, total plasma phenytoin levels underrepresent the concentration of unbound, pharmacologically active drug.
Tissue Binding
Some drugs are found in certain tissues at concentrations much higher than concentrations in plasma. To some extent, this may be a consequence of partitioning related to the physical-chemical properties of the drug; a highly lipophilic compound, for example, accumulates in lipidic tissues in a manner roughly proportional to its partition coefficient between oil and water. In some cases, drugs may be “sequestered” in tissues as a result of binding with proteins and other macromolecules. Because the bound drug is not available to interact with receptor sites, the total drug concentration in a given tissue might not provide a reliable estimate of the drug concentration at sites of action within the same tissue.
Tissue Blood Flow and Drug Distribution
After drug administration, distribution phenomena may take a long time to be completed. Initially, distribution tends to be confined primarily to highly perfused organs, such as the heart, liver, kidneys, and brain, which receive a large fraction of cardiac output. Subsequently, the drug redistributes gradually throughout the organism, and distribution can be considered complete only when the plasma-to-tissue concentration ratio has reached equilibrium in all tissues.
A good example of the clinical implications of redistribution phenomena is provided by thiopental, a compound sometimes used in the management of refractory status epilepticus. After a single injection of thiopental, the drug is transferred largely to the brain, where it penetrates rapidly because of its high lipophilicity. Other lipid-rich tissues (e.g., adipose tissue) initially receive little exposure to the drug because their perfusion is low compared with that of the brain. During the subsequent minutes, however, thiopental is redistributed gradually out of the brain into less perfused tissues, which tend to accumulate it at high concentrations.19 Redistribution is mainly responsible for termination of action after a single dose, and it explains the apparent discrepancy between the short duration of thiopental-induced anesthesia (20–30 minutes) and the relatively long elimination half-life of the drug (about 9 hours). The situation is completely different when thiopental is administered by continuous infusion over several days. Under these circumstances, drug stores in poorly perfused tissues become gradually saturated. When treatment is discontinued, redistribution no longer occurs, and the termination of drug action is then determined by hepatic elimination, which explains the long delay before consciousness is recovered.
Blood–Brain Barrier
The anatomic configuration of the cerebral circulation represents a barrier to the entry of certain drugs into the brain and cerebrospinal fluid. As a general rule, the ability of a compound to diffuse across the blood–brain barrier is directly proportional to its lipid solubility. A number of poorly lipid-soluble drugs, however, may be transported in or out of the brain by selective uptake mechanisms. For example, l-dopa is transported into the central nervous system by a carrier-mediated process, whereas penicillin and salicylic acid are transported out of the brain by a different system of carriers. When carriers are involved, there is possibility of competition by other substrates at the site of transport; some amino acids, for example, compete with l-dopa for entry into the brain, whereas probenecid may block the uptake of penicillin from the cerebrospinal fluid by a similar mechanism.
The kinetics of penetration of antiepileptic drugs across the blood–brain barrier has been investigated in the anesthetized dog.44 The fastest rates of entry were recorded for clonazepam, N-desmethyldiazepam, diazepam, and ethosuximide, whereas valproic acid, phenytoin, phenobarbitone, carbamazepine, and particularly primidone crossed the blood–brain barrier at a much slower rate (Table 2). Ionization did not appear to be a rate-limiting factor in brain penetration because all drugs except for valproic acid are mostly nonionized at physiologic pH. Furthermore, no correlation was found between rate of brain penetration and plasma protein binding, but for most drugs the ratio of cerebrospinal fluid to total plasma concentration at equilibrium was almost equal to the unbound fraction in plasma. Rate of entry into the cerebrospinal fluid correlated with lipid solubility as assessed by the benzene/buffer distribution ratio. Valproic acid showed unique features because it crossed the blood–brain barrier relatively rapidly despite its very low lipid solubility. These data suggest that the brain penetration of valproic acid may be regulated by an active transport process. There is also evidence that valproic acid may be transported out of the cerebrospinal fluid by an active uptake system localized in the choroid plexus, an observation that may explain the very low concentrations of the drug (relative to plasma concentrations) in the brains of patients receiving chronic treatment.89
In recent years, the mechanisms affecting access of drugs to the brain have been extensively investigated. Evidence has accumulated that, for many drugs, access to brain is influenced by the expression of efflux transporters, including P-glycoprotein, located in the cerebral capillary endothelial cells and in other cells, for example, astrocytes.45 These transporters play a key role in reducing the penetration of substrates across barriers where they are expressed, including the blood–brain barrier, the blood cerebrospinal fluid (CSF)–barrier, and the blood–inner ear barrier.39 Studies have suggested that several anticonvulsants, including phenytoin, phenobarbital, carbamazepine,
lamotrigine, gabapentin, topiramate, and felbamate, are substrates of P-glycoprotein and other transporters, although evidence is not univocal for all drugs.39 It has also been suggested that overexpression of these transporters may be a cause of drug resistance by limiting access of drugs to their site of action in neurons.39,86
lamotrigine, gabapentin, topiramate, and felbamate, are substrates of P-glycoprotein and other transporters, although evidence is not univocal for all drugs.39 It has also been suggested that overexpression of these transporters may be a cause of drug resistance by limiting access of drugs to their site of action in neurons.39,86
Table 2 Rates of penetration of different antiepileptic drugs from plasma into cerebrospinal fluid in anesthetized dogsa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The permeability of the blood–brain barrier is influenced by physiologic and pathologic factors. The increased sensitivity of neonates to some centrally acting drugs may be related, at least in part, to enhanced permeability of the blood–brain barrier during early stages of development. Many diseases can affect the integrity of the barrier. Disruption of the blood–brain barrier, resulting in increased brain exposure to valproic acid, may explain the occurrence of severe valproate encephalopathy within days of supratentorial surgery in some patients.40
Selective Drug Delivery to the Brain
The existence of the blood–brain barrier can be exploited by developing chemical systems designed to ensure preferential drug delivery to the brain. The best known of these systems is based on the dihydropyridinepyridinium salt–type redox molecular carrier.79 In brief, the drug is conjugated with a 1,4-dihydropyridine moiety, resulting in a lipophilic complex that readily penetrates all tissues, including the brain. Thereafter, the complex undergoes quick oxidation to a highly polar quaternary pyridinium salt that is rapidly eliminated from the periphery but remains “locked” in the brain because of the impermeability of the blood–brain barrier. Over time, the active drug is released from the oxidized complex by enzymatic hydrolysis and becomes available to exert its pharmacologic action. The small, polar targeter released by the process of hydrolysis is readily expelled from the brain through an active transport mechanism. Because the system allows selective drug delivery to the brain, peripheral toxic effects can be minimized. Central tolerability may also be improved because of the slow and sustained delivery of the active principle at the site of action.
Experimental delivery systems based on the dihydropyridinepyridinium salt–type redox molecular carrier have been developed for a variety of antiepileptic drugs, including phe-nytoin, valproic acid, and stiripentol. Although at present the practical applicability of these systems is limited by the instability of dihydropyridine moiety in gastric fluids, innovative formulations for clinical use may become available in the future.
Placental Barrier
The use of the term barrier to indicate the anatomic structures regulating drug exchanges between mother and fetus is improper. Unlike the blood–brain barrier, generally the placenta does not provide an efficient obstacle to drug penetration. Although lipophilic drugs may enter the fetal circulation more readily than water-soluble compounds, from a practical point of view the fetus can be considered as subject to exposure to all drugs taken by the mother. For gabapentin, drug concentrations have been reported to be almost twice as high in the fetal circulation than in the mother, possibly due to existence of an active transplacental transport.57
Distribution Into Breast Milk
The distribution of drugs into breast milk is determined by a number of variables, the most important being the nonionized fraction of the drug in plasma and milk, lipid solubility, and the binding of the drug to plasma and milk proteins.102 Drug exposure in the breast-fed infant, however, depends not only on the concentration of the drug in milk and the amount of milk ingested, but also on the infant’s ability to absorb and clear efficiently the ingested drug.
All anticonvulsants pass into breast milk and the average milk-to-plasma concentration ratio ranges from about 0.7 to 1.4 for gabapentin and levetiracetam to 0.01 to 0.1 for valproic acid, which is hydrophilic and highly protein bound.30,57,60 As a general rule, the presence of antiepileptic drugs in breast milk is not considered to be a contraindication to breast-feeding, although for some medications, particularly lamotrigine, primidone, phenobarbital, ethosuximide, and, to a lesser extent, carbamazepine and benzodiazepines, intake through breast milk may result in appreciable drug concentrations in the infant and the need to monitor for possible side effects. Because felbamate-induced hematological toxicity may be dose independent, use of this drug during breast-feeding has been discouraged.
Elimination
The most important routes of drug elimination involve direct excretion, mostly into urine, and biotransformation with subsequent excretion of breakdown products in urine and/or bile. The extensive interindividual variability of these processes is largely responsible for the wide differences in plasma drug levels observed in patients receiving the same dose. Therefore, a correct understanding of the factors that may affect drug elimination is crucial for rational prescribing.
Pharmacokinetic Concepts
Drug Clearance
The pharmacokinetic parameter that best describes the efficiency of elimination processes is the clearance, defined as the virtual volume of blood (or plasma) from which the drug is entirely removed per unit of time.101 When a drug is eliminated exclusively by the kidneys in unchanged form, blood clearance equals renal clearance. When metabolism contributes to elimination, blood clearance is equal to the sum of renal clearance and metabolic clearance. Within any organ involved in drug elimination, the efficiency of the elimination process can be defined according to the formula
where CL1 is the contribution of that organ to the total clearance (e.g., in the case of the kidney, CL1 equals renal clearance), Q is the blood flow through the organ, and E is the extraction ratio (i.e., the fraction of drug passing through the organ that is removed as a result of the elimination process). If a drug is totally removed during a single passage, E is equal to 1 and CL1 becomes a direct measure of the blood flow through the organ.
From inspection of Equation (1), it is clear that drug clearance is influenced not only by the capacity of the organ to remove the drug efficiently from the bloodstream, but also by the rate of drug delivery to the site of elimination. In this respect, it is useful to differentiate between low- and high-clearance drugs.
In the case of low-clearance drugs, the efficiency of the elimination processes is relatively low, and protein-bound drug cannot be removed by the eliminating organ. The clearance of these drugs is restricted by the degree of binding to plasma proteins; a reduction in plasma protein binding, by increasing the availability of unbound molecules available for removal, will result in a corresponding increase in clearance. Conversely, the clearance of these drugs will not be affected by blood flow because any increase in rate of delivery of the drug to the eliminating organ due to increased blood flow will be offset by the organ’s inability to maintain the efficiency of drug removal (i.e., an increase in blood flow will be accompanied by a corresponding decrease in extraction ratio, and clearance will remain unchanged). For these reasons, low-clearance drugs are said to exhibit restrictive, flow-independent elimination.101
The situation is reversed in the case of high-clearance drugs. These compounds are removed from the circulation so efficiently that protein-bound drug can also be eliminated during a single passage through the organ. The clearance of these drugs is nonrestrictive because plasma protein binding does not restrict the removal process but may actually facilitate it by increasing the delivery of the drug to the site of elimination. The clearance of these drugs is also flow dependent. In fact, in this case the eliminating organ can cope with an increase in blood flow without losing its extraction capacity, resulting in a direct relationship between blood flow and drug clearance. When the organ responsible for drug removal is the intestine or the liver, a third characteristic of high-clearance drugs is a prominent first-pass effect after oral administration. This is explained by the fact that the efficient elimination (i.e., a high extraction ratio) leads to removal of a significant fraction of the dose before the drug reaches the systemic circulation.
Elimination Half-life
An additional pharmacokinetic parameter used to describe drug elimination is the terminal half-life (or, more simply, half-life), which can be defined as the time taken for the plasma concentration to fall by one half during the terminal (postdistributive) elimination phase. The half-life is a useful parameter because it provides a direct measure of the rate of decline of drug concentration in plasma after discontinuation of therapy. In pharmacokinetic terms, however, the half-life is a hybrid parameter, determined not only by the efficiency of the elimination processes (i.e., drug clearance), but also by the volume of distribution. This can be understood easily by considering that after removal of any given amount of drug per unit of time, the percentage of reduction in plasma drug concentration primarily depends on the total drug stores in the body. If body stores are large (i.e., volume of distribution is large), the fall in concentration resulting from the removal will be minor. Conversely, if body stores are small, removal of the same amount of drug will have a much greater impact on plasma concentration.
Based on the foregoing considerations, it is clear that a change in half-life does not always reflect a change in the efficiency of the elimination process (i.e., drug clearance) but may be secondary to an alteration in volume of distribution. For example, the marked prolongation in the half-life of diazepam in the elderly is determined largely, although not exclusively, by an increase in volume of distribution.35 The differentiation between clearance and half-life has important practical implications. In fact, mean plasma drug concentrations at steady state during long-term dosing are determined exclusively by bioavailability and clearance and are unaffected by half-life. Conversely, the half-life determines the time required to reach steady state (about four to five half-lives) as well as the degree of fluctuation in plasma levels during a dosing interval.
Biotransformation
Most drugs are too lipid soluble to be excreted efficiently by the kidneys, and after being filtered by the renal glomeruli, they are almost completely reabsorbed by diffusion from the tubular epithelium.77 Elimination of these compounds depends primarily on their conversion to polar and water-soluble metabolites that can be excreted more rapidly in urine. In general, biotransformation products are inactive or less active than the parent drug, although there are numerous examples of metabolites retaining pharmacologic activity and contributing significantly to the production of therapeutic or toxic effects. Among antiepileptic drugs, those converted at least in part to active metabolites include clobazam, diazepam, carbamazepine, primidone, fosphenytoin, and oxcarbazepine (Table 3). Fosphenytoin and oxcarbazepine can be regarded as prodrugs because their effects are mediated virtually entirely by their active metabolites—phenytoin in the case of fosphenytoin, and 10-hydroxycarbazepine in the case of oxcarbazepine.
Almost invariably, the reactions involved in drug meta-bolism are catalyzed by enzyme systems and often exhibit a considerable degree of stereoselectivity. The most important enzyme systems are located in the smooth endoplasmic reticulum (microsomes) of hepatic cells, but significant levels of drug-metabolizing activity can be detected in the microsomes and the cytosol of other tissues, including the gastrointestinal mucosa, kidney, lung, blood, and brain.
The main metabolic pathways involved in drug metabolism include oxidation, reduction, hydrolysis, and conjugation. The first three are collectively known as phase I reactions, whereas conjugations are generally referred to as phase II reactions. In phase I reactions, functional groups such as —OH, —COOH, and —NH2 are introduced into the drug molecule. In phase II reactions, functional groups already present in the molecule or introduced in the previous step are conjugated with an endogenous substrate, such as glucuronic acid, acetic acid, or inorganic sulfate, resulting in the generation of water-soluble metabolites that are readily excreted in urine.
Table 3 Main routes of elimination of antiepileptic drugs | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


