aPercentage of incidentally discovered adrenal masses with adrenal hyperfunction or cancer.
Modified from Young WF Jr. Clinical practice: The incidentally discovered adrenal mass. N Engl J Med 2007;356:601–610.
Evaluation for Malignancy
Potential malignancy is an overriding concern. Adrenocortical carcinoma is detected in 4.7% and metastatic cancer in 2.5% (Young, 2007b).
As shown in Table 12-2, the size of the mass and its appearance on CT (or MRI)—the imaging phenotype—are the two key indicators of malignancy (Young, 2007b).
TABLE 12-2 Typical Imaging Features (Phenotype) of Incidental Adrenal Masses
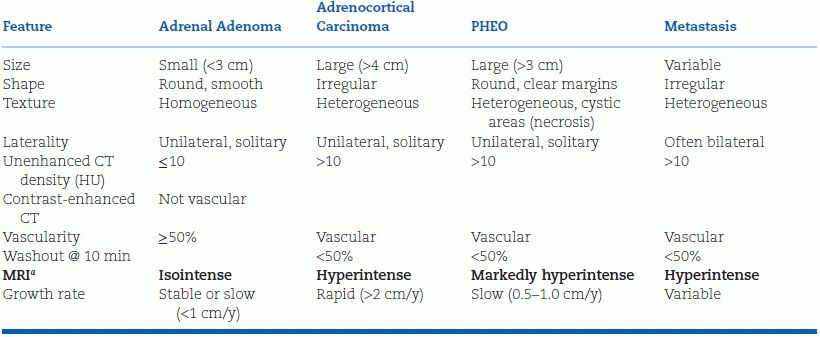
aRelative to liver on T2-weighted imaging.
Modified from Young WF Jr. The incidentally discovered adrenal mass. N Engl J Med 2007;356:601–610.
Size
Adrenocortical carcinomas typically are large; 90% are at least 4 cm in diameter. Among the patients with adrenal incidentaloma greater than 4 cm in diameter, one in four will have adrenocortical carcinoma (Young, 2007b). The smaller the carcinoma at the time of resection, the lower will be the tumor stage and the better the prognosis.
Imaging Phenotype
Most adrenal incidentalomas are discovered unexpectedly on contrast-enhanced abdominal or chest CT scans that often are not technically optimal for adrenal imaging (Terzolo et al., 2011). Thus, an adrenal protocol scan should be used to further assess the incidentaloma: contiguous 3- to 5-mm-thick CT slices should be obtained without contrast, 1 minute after intravenous injection of contrast media, and 10 to 15 minutes later (Zeiger et al., 2011).
In adenomas, the cytosol typically is laden with fat, yielding characteristic features on CT and MRI (Zeiger et al., 2011). On noncontrast CT, benign adenomas typically have a low attenuation value measured in Hounsfield units (HU). If the noncontrast CT attenuation value is less than 10 HU, the patient can be assured that the tumor is a benign lipid-rich adenoma (Zeiger et al., 2011). On T2-weighted MRI, adenomas are isointense to the liver or spleen. On chemical shift MRI, signal loss occurs on the out-of-phase images. However, up to 30% of adenomas do not contain much fat and thus cannot be distinguished from malignancy or pheochromocytoma (PHEO) by noncontrast CT or MRI. If washout of the contrast material is greater than 50% complete 10 minutes after injection, the patient can be reassured (with virtually 100% sensitivity and specificity) that this is a benign adenoma (Terzolo et al., 2011). Washout is slower with PHEO or adrenal malignancy. If malignancy is still uncertain, a PET scan with 18F-FDG has a very high sensitivity (93% to 100%) and specificity (80% to 100%) for identifying malignant lesions, which show excessive uptake of glucose (Terzolo et al., 2011).
Metastases
Primary cancers that commonly metastasize to the adrenals are carcinomas of the lung, kidney, and gastrointestinal (GI) tract. Metastases tend to cause bilateral adrenal masses, and the primary tumor often has been discovered before the adrenal incidentaloma(s) (Zeiger et al., 2011).
Evaluation for Hyperfunction
Table 12-3 lists the screening procedures and the confirmatory tests for adrenal hyperfunction, i.e., autonomous production by an adrenal tumor of cortisol, catecholamines, or aldosterone. Among 3,868 patients with adrenal incidentaloma in 26 series, biochemical evidence of subclinical Cushing syndrome was found in 7.9%, PHEO in 5.6%, and primary aldosteronism in 1.2% (Barzon et al., 2003). More recent data suggest that some degree of increased cortisol secretion occurs in up to 33% (Kmietowicz, 2014).
TABLE 12-3 Laboratory Evaluation of an Incidental Adrenal Mass

Modified from Young WF Jr. Clinical practice: The incidentally discovered adrenal mass. N Engl J Med 2007;356:601–610.
One-quarter to three-quarters of adrenocortical carcinomas are hormonally active. Cosecretion of cortisol and androgens is the most common pattern and is highly suggestive of adrenocortical carcinoma (Libe et al., 2007).
Subclinical Cushing Syndrome
Subclinical Cushing syndrome needs to be considered when an adrenal incidentaloma is accompanied by subtle clinical signs of hypercortisolism. Patients can have hypertension, central obesity, diabetes, fatigue, and easy bruising; however, they do not have the wide nonblanching purple striae or other signs of full-blown Cushing syndrome (Terzolo et al., 2011; Zeiger et al., 2011). Thus, the clinical picture is hard to distinguish from the garden-variety metabolic syndrome—except imaging studies have uncovered an adrenal mass.
To improve the sensitivity of the 1 mg overnight dexamethasone suppression test, a lower-than-standard cutoff—1.8 mg/dL rather than 5 mg/dL—should be used for an abnormally elevated 8 a.m. cortisol value; to avoid false positives, the minimally elevated value should be confirmed repeatedly and accompanied by feedback suppression of adrenocorticotropic hormone (ACTH), additional evidence for autonomous adrenal overproduction of cortisol (Terzolo et al., 2011; Zeiger et al., 2011).
Subclinical Cushing syndrome constitutes a novel cardiovascular risk factor as shown by a recent retrospective study of 198 patients with an incidental adrenal mass and no overt disease were followed for an average of 7.5 years with cortisol levels measured after 1 mg dexamethasone suppression testing (Di Dalmazi et al., 2014). Of these, 114 patients (58%) had stable nonsecreting adrenal masses (postdexamethasone plasma [cortisol] < 50 nmol/L), 61 patients (30%) had either a stable intermediate phenotype ([cortisol] 50 to 138 nmol/L) or subclinical Cushing syndrome ([cortisol] > 138 nmol/L), and 23 patients (12%) who had a progressive pattern of increasing cortisol secretion. Compared with patients with stable nonsecreting masses, the rate of cardiovascular events and death was higher in patients with intermediate disease or subclinical Cushing syndrome (6.7% vs. 16.7%) and those with worsening secretion (6.7% vs. 28.4%). An increase in cortisol secretion during the study was an independent risk factor for having a new cardiovascular event.
Subclinical Cushing’s syndrome is particularly common with adrenal incidentalomas larger than 2.4 cm (Morelli et al., 2014) and with bilateral adrenal incidentalomas, where the prevalence is (35% vs. 18% with unilateral incidentaloma) (Vassilatou et al., 2014).
Clinically Silent PHEO
In the Mayo Clinic series, approximately 5% of adrenal incidentalomas turned out to be PHEOs (Young, 2007b). The likelihood of discovering a PHEO is approximately 25-fold higher when hypertension is accompanied by an adrenal incidentaloma than in the general hypertensive population. Currently, more than half of all PHEOs are discovered incidentally; of these, one-half are accompanied by neither hypertension nor other classical clinical features (Zeiger et al., 2011). The diagnosis is based on biochemistry, i.e., demonstration of catecholamine oversecretion by the adrenal chromaffin cells (see below).
Primary Aldosteronism
In the Mayo Clinic series, 1% of incidentalomas turned out to be aldosterone-producing adenomas (Young, 2007b). The best screening test is a blood sample for an elevated serum aldosterone level and suppressed plasma renin activity (see Chapter 11).
Management
Figure 12-1 is an algorithm for evaluating the patient with an adrenal incidentaloma.
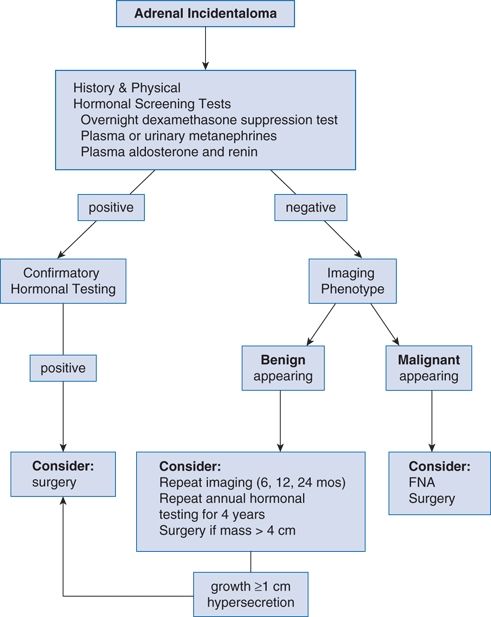
FIGURE 12-1 Algorithm for diagnostic evaluation of an incidental adrenal mass. FNA, fine needle aspiration. (Modified from Young WF Jr. Clinical practice: The incidentally discovered adrenal mass. N Engl J Med 2007b;356:601–610.)
Initial hormonal evaluation should include the following three screening tests: (a) an overnight dexamethasone (1 mg) suppression test for subclinical Cushing syndrome, (b) measurement of plasma free metanephrines or of fractionated metanephrines and catecholamines in a 24-hour urine specimen for PHEO, and (c) measurement of serum aldosterone and plasma renin activity for primary aldosteronism.
Even clinically silent PHEOs can trigger lethal hypertensive crisis and therefore should be resected after preoperative adrenergic blockade. Aldosterone-producing adenoma is an indication for laparoscopic adrenalectomy if accompanied by hypertension or hypokalemia.
The approach to subclinical Cushing syndrome is a work in progress. In an uncontrolled study of nine patients with this diagnosis, unilateral adrenalectomy ameliorated hypertension in six patients and reduced supraclavicular fat pads and other clinical features in all nine (Mitchell et al., 2007). These provocative findings indicate the need for a large multicenter trial.
For now, adrenalectomy for subclinical Cushing syndrome should be considered in younger patients (under age 40) with recent onset or deterioration of hypertension, diabetes, and other clinical features of hypercortisolism. In middle-aged or older patients, a large adrenal mass favors resection. Most cortisol-producing adenomas are 2.5 cm or larger. If surgery is undertaken, glucocorticoid therapy should be administered to avoid perioperative adrenal crisis.
Adrenalectomy is indicated when the radiologic appearance is suspicious for adrenal carcinoma, unless there are extenuating clinical circumstances related to advanced age and comorbidity. If an adrenal mass is ≥6 m in diameter, it needs to be resected. If an adrenal mass is 4 to 6 cm in diameter, the patient’s age and imaging phenotype should be considered. Before age 30, adrenal incidentalomas are so rare that even a mass ≤4 cm merits consideration for resection, particularly if the imaging phenotype is suspicious (Young, 2007b).
Fine needle aspiration (FNA) biopsy is rarely needed to exclude malignancy because the imaging characteristics are much more predictive (Terzolo et al., 2011). FNA is used mainly to exclude metastatic disease or infection (e.g., adrenal TB). A PHEO must be excluded first by biochemistry because FNA of a PHEO can trigger hypertensive crisis.
If the mass has a benign appearance on CT or MRI and the initial hormonal studies are negative, imaging studies should be repeated every 6 months for up to 2 years and adrenal function studies should be repeated yearly for up to 4 years (Terzolo et al., 2011). Only then can the patient be reassured that there is little chance of further trouble.
OVERVIEW OF ADRENAL HYPERTENSION
As stated above, every hypertensive patient with an incidental adrenal mass will merit a workup for adrenal hypertension. Indeed, all hypertensive patients merit consideration of a potential adrenal cause but, without a known adrenal mass, only a small percentage will need an evaluation. These adrenal diseases are rare and produce nonspecific signs and symptoms. Many patients with primary hypertension have recurrent spells suggesting PHEO, hypokalemia suggesting primary aldosteronism, and central obesity suggesting subclinical Cushing syndrome. Most will turn out to have normal adrenal function.
Although uncommon, the diagnosis of adrenal hypertension can lead to surgical cure or highly effective targeted drug therapy. Removing a hyperfunctioning adrenal tumor or blocking the effects of hormonal excess on target tissues may be the only way to adequately control the hypertension and protect the patient from rampant target organ damage and premature death. This is particularly the case with PHEO.
PHEOCHROMOCYTOMA AND PARAGANGLIOMA
PHEOs are catecholamine-secreting tumors of adrenal chromaffin cells. Paragangliomas are extra-adrenal tumors of sympathetic or vagal ganglion cells. The odds that a paraganglioma will secrete catecholamines vary by location, as shown in Table 12-4. Catecholamine-secreting paragangliomas—often called “extra-adrenal PHEOs”—are located mainly in the abdomen or pelvis. Nonsecreting vagal paragangliomas are located mainly in the head and neck, most frequently involving the glomus cells of the carotid body. While the term PHEO often is used to include adrenal PHEOs and extra-adrenal paragangliomas, they differ not only in location but also in clinical presentation, genetic underpinning, and malignant potential (Waguespack et al., 2010).
TABLE 12-4 Location of Paraganglioma

Modified from Young WF Jr, Abboud AL. Editorial: Paraganglioma—all in the family. J Clin Endocrinol Metab 2006;91:790–792.
Most PHEOs and paragangliomas secrete both norepinephrine (NE) and epinephrine (EPI), with NE predominating. Some primarily secrete EPI, and an occasional tumor will secrete only dopamine. When correctly diagnosed and treated, most PHEOs are curable. When undiagnosed or improperly treated, they can be fatal.
PHEOs often go unrecognized (Jones et al., 2012). In an autopsy series at Mayo Clinic, only 13 of 54 autopsy-proven PHEOs had been diagnosed during life (Young, 2007a). The remaining 41 undiagnosed PHEOs caused 30 deaths. They also are overdiagnosed due to false-positive test results (Yu & Wei, 2010). In contrast, when PHEO is diagnosed and managed by a highly experienced clinical team, the tumors can be successfully resected with minimal perioperative mortality (Darr et al., 2012; Young, 2007a).
Prevalence
PHEOs are rare. The estimated prevalence is less than 0.2% among unselected patients with hypertension but 5% among those with adrenal incidentaloma (Barzon et al., 2003; Young, 2007a). Because PHEOs are so rare and can cause lethal paroxysms, the clinician needs a high index of suspicion and a systematic approach to screening, localization, and surgery (Fig. 12-2).
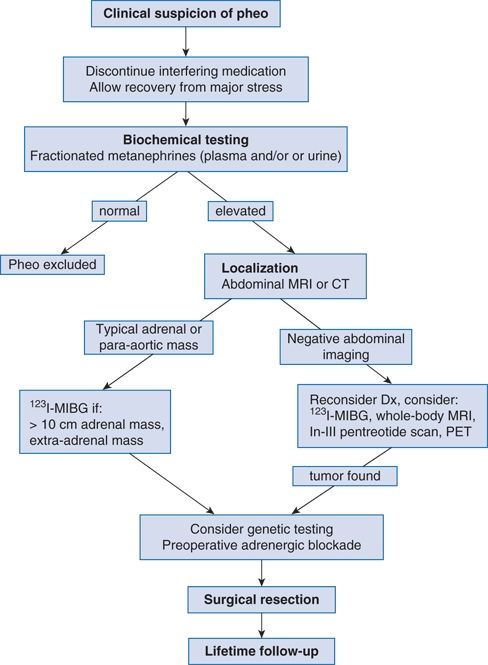
FIGURE 12-2 Algorithm for diagnostic evaluation of a suspected pheochromocytoma. PHEO, adrenal pheochromocytoma or extra-adrenal paraganglioma; In, indium. (Modified from Young WF Jr. Adrenal causes of hypertension: Pheochromocytoma and primary aldosteronism. Rev Endocr Metab Disord 2007a;8:309–320.)
Clinical Features
Table 12-5 lists the varied clinical features of PHEO. Table 12-6 lists the long differential diagnosis.
TABLE 12-5 Signs and Symptoms of PHEO
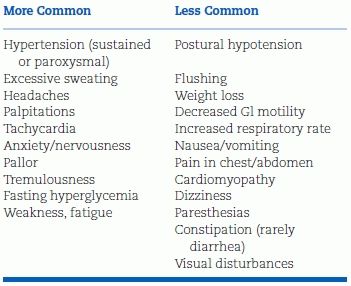
Modified from Pacak K. Preoperative management of the PHEO patient. J Clin Endocrinol Metab 2007;92:4069–4079.
TABLE 12-6 Differential Diagnosis of PHEO-Like Spells

Hyperadrenergic spells are the classic clinical presentation of PHEO. Excessive α- and β-adrenergic stimulation of the cardiovascular system produces the five “Ps” of the paroxysm (Young WF Jr, Personal communication, 2007c):
- Paroxysmal hypertension
- Pounding headache
- Perspiration (often diffuse)
- Palpitations
- Pallor (not flushing)
Flushing is less common than pallor because NE—the dominant catecholamine—is a potent vasoconstrictor. Diabetes and weight loss are other signs of the hyperadrenergic state. Some patients are asymptomatic, some are normotensive, and others have symptoms due to concomitant conditions.
Paroxysmal Hypertension
The paroxysms represent the classic picture of the disease, but exclusively paroxysmal hypertension with intervening normotension is rare. Patients can have sustained hypertension with or without superimposed paroxysms. The paroxysms can be triggered by mechanical compression of the tumor (by exercise, upright posture, bending over, urination, defecation, an enema, palpation of the abdomen, or a pregnant uterus), injection of chemicals (anesthetic agents or radiology contrast material), drugs that stimulate catecholamine synthesis (glucocorticoids) and secretion (histamine, opiates, or nicotine), psychiatric drugs that inhibit biogenic amine reuptake transporters (tricyclic antidepressants, selective NE reuptake blockers, etc.), and β-blockers, which leave α-adrenergic receptors relatively unopposed. PHEO crisis can be induced by glucocorticoids, including ACTH, methylprednisolone, and high-dose (but not 1-mg low-dose) dexamethasone suppression testing (Rosas et al., 2008). The paroxysm does not occur immediately but rather 5 to 36 hours after the glucocorticoid administration and is caused by tumor necrosis. Paroxysms also may occur without provocation from spontaneous tumor necrosis.
Among individual patients, paroxysms vary in frequency, duration, severity, and associated symptoms. They may occur many times per day or only every few months. PHEO paroxysms often are misdiagnosed as panic attacks. Patients may describe tightness in the abdomen rising into the chest or head, anxiety, tremors, sweating, palpitations, and weakness.
Cardiac Manifestations of PHEO
In the Cedars-Sinai series, 12% of PHEOs presented without paroxysmal hypertension but rather with acute coronary syndrome or catecholamine-induced cardiomyopathy with acute heart failure (inverted Takotsubo cardiomyopathy) (Yu et al., 2012a). PHEO also may present as monomorphic ventricular tachycardia (Park et al., 2012). A high index of suspicion is needed to make the diagnosis because PHEO is far rarer than the common forms of adult heart disease it mimics. Failure to consider PHEO or especially paraganglioma in the differential diagnosis can lead to unnecessary invasive procedures, including cardiac transplantation (Yu et al., 2012a). Compared with PHEO patients presenting with either adrenal incidentaloma or paroxysmal hypertension but without cardiac manifestations, those presenting with cardiac complications have large tumors (often >6 cm), markedly elevated plasma normetanephrine (NMN) levels, mildly depressed left ventricular ejection fractions (LVEFs), and prolonged QT intervals (Table 12-7) (Yu et al., 2012a). They may also present with an embolic stroke due to left ventricular thrombus (Buchbinder et al., 2009). Thus, in the absence of paroxysmal hypertension, PHEO should be considered in an adult with no convincing etiology of heart failure, acute coronary syndrome, or tachyarrhythmia with or without an adrenal mass (the latter due to paraganglioma). Spot plasma metanephrines will make the biochemical diagnosis.
TABLE 12-7 Characteristics of Patients with PHEO Presenting with or without Cardiac Complications
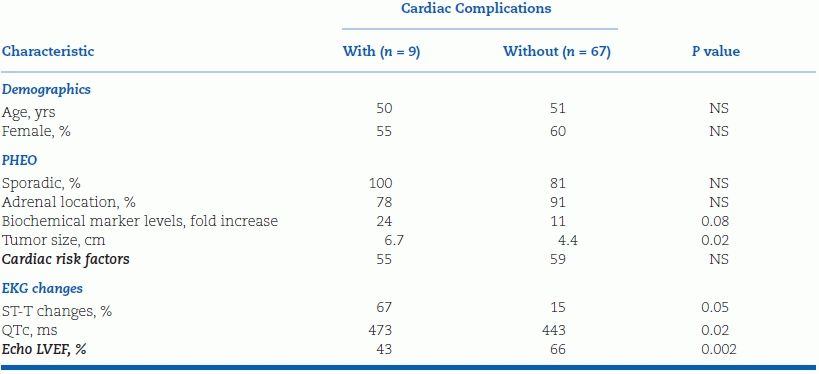
Modified from Yu R, Nissen NN, Bannykh SI. Cardiac complications as initial manifestation of pheochromocytoma: Frequency, outcome, and predictors. Endocr Pract 2012;18:483–492.
Hypotension
Predominantly EPI-secreting PHEOs can present as hypotension, as cyclic attacks of hypertension alternating with hypotension, and as acute coronary syndromes: diffuse ST-segment depression, chest pain, nausea/vomiting, and diaphoresis (Darr et al., 2012). They can cause cardiogenic shock from catecholamine-induced cardiomyopathy. Profound hypotension may occur with spontaneous tumor necrosis or with α-blocker administration during the preoperative preparation for PHEO surgery. More commonly, patients have modest postural hypotension with tachycardia and dizziness. The postural hypotension indicates hypovolemia, which is a characteristic of PHEO that has never been adequately explained. In an untreated young hypertensive, postural hypotension and tachycardia may be a clue to the presence of a PHEO.
Less Common Presentations
PHEOs also can present as an acute abdomen (from spontaneous tumor rupture), sudden death after minor abdominal trauma, lactic acidosis, or high fever and encephalopathy. Paragangliomas of the urinary bladder can paroxysmal attacks of micturition syncope or painless hematuria (Beilan et al., 2013); they tend to occur in rather young adults and most can be cured by partial cystectomy.
The Revised Rule of 10s
Conventional teaching was that 10% of PHEOs are extra-adrenal (i.e., secretory paragangliomas), 10% occur in children, 10% are bilateral, 10% recur, 10% are malignant, 10% are discovered incidentally, and 10% are familial. Now, up to 70% are discovered incidentally (Darr et al., 2012; Yu et al., 2009) and over 30% are due to inherited germ line mutations (i.e., mutations occurring in all the cells of the body) (Karasek et al., 2013).
Familial PHEO and Paraganglioma
PHEOs and paragangliomas can occur sporadically or they can be inherited as autosomal dominant traits alone or as part of one of several syndromes listed below. As recently reviewed by Karasek and coworkers at the NIH (Karasek et al., 2013), the known disease-causing genes are as follows:
- RET (rearranged during transfection), a protooncogene associated with Multiple Endocrine Neoplasia (MEN) type 2A (PHEO, medullary carcinoma of the thyroid, hyperparathyroidism) or type 2B (PHEO, medullary carcinoma of the thyroid, mucosal neuromas, thickened corneal nerves, intestinal ganglioneuromatosis, slender facies).
- VHL, a tumor suppressor gene associated with von Hippel-Lindau disease of PHEO (often bilateral), retinal and cerebellar angiomas, renal and pancreatic cysts, and renal cell carcinoma.
- NF-1, associated with neurofibromatosis.
- SDHD, succinate dehydrogenase (mitochondrial complex II) gene subunit D that predisposes to familial paraganglioma.
- SDHB, succinate dehydrogenase gene subunit B that also predisposes to familial paraganglioma.
- SDHC, succinate dehydrogenase gene subunit C that mainly predisposes to nonsecretory vagal paragangliomas of the head and the neck.
- SDHA, succinate dehydrogenase A subunit, mutations are associated mainly with abdominal paragangliomas (rarely adrenal PHEOs).
- SDHAF2, succinate dehydrogenase A F2 subunit, mutations are associated with head and neck paragangliomas and paternal transmission.
- TMEM-127, transmembrane protein 127 gene mutations are associated with mainly nonmalignant adrenal PHEOs.
- MAX, protein MAX (also known as MYC_associated factor X), interacts with other transcription factors forming a network that regulates cell proliferation, differentiation, and apoptosis. MAX behaves as a tumor suppressor gene; thus, 2/3 of MAX-associated PHEOs are often bilateral and 25% are malignant.
Differing Phenotypes of MEN2 and VHL Syndrome
Patients with MEN2 are more likely to suffer from paroxysmal hypertension because MEN2 tumors mainly secrete NE whereas VHL tumors mainly secrete EPI. Compared with VHL tumors, MEN2 tumors have higher expression of both tyrosine hydroxylase, the rate-limiting enzyme in catecholamine synthesis, and phenylethanolamine N-methyltransferase (PNMT), the enzyme that converts NE to EPI.
The first description of PHEO from 1886 was, in fact, a case of MEN2 syndrome (Neumann et al., 2007). The patient, an 18-year-old German girl named Mina Roll, presented with classic symptoms of PHEO crisis and, at autopsy, was found to have vascular bilateral adrenal tumors that stained brown with chromate fixative (hence the name “PHEO chrom” which means “chromate brown”). With a careful piece of detective work 120 years later, the geneticists searched the European-American Pheochromocytoma Registry to find four living family members with a germ line mutation in the RET gene, thus establishing the diagnosis of MEN2. These and other family members had PHEOs and/or medullary carcinoma of the thyroid, the latter explaining the goiter in Mina Roll’s original autopsy report (Neumann et al., 2007). Early clinical recognition of MEN2 syndrome is particularly important because medullary carcinoma of the thyroid—present in most of these patients—poses a major cause of death and thus merits immediate surgical removal.
Neurofibromatosis
PHEO occurs in only 1% of patients with neurofibromatosis type 1. As of 2006, neurofibromatosis accounted for only 25 cases of the 565 total PHEO cases in the European-American Pheochromocytoma Registry (Bausch et al., 2006). By comparison, von Hippel-Lindau syndrome accounted for 75 cases, paraganglioma syndromes for 54 cases, and sporadic PHEO for 380 cases. All 25 of 25 patients with neurofibromatosis had adrenal PHEOs and, of these, three (12%) had metastatic disease.
Familial Paraganglioma
Discovered only in 2000, the familial paraganglioma syndromes are inherited as autosomal dominant traits (Darr et al., 2012; Karasek et al., 2013). Of note, the SDHD mutation is characterized by maternal imprinting, meaning that the disease can only be inherited from one’s father, emphasizing the importance of a detailed family history. The SDHB mutation is characterized by a high risk of malignancy including metastatic paraganglioma, renal cell carcinoma, and papillary carcinoma of the thyroid. The SDHC mutation is mainly associated with nonsecreting vagal paragangliomas (most often carotid body tumors) of the head and neck. Production of 3-methoxytyramine is associated with the presence of an underlying SDHB mutation and may be a viable biomarker of malignancy (van Berkel et al., 2014).
Tumorigenesis may involve (a) the failure of developmental apoptosis and/or (b) pseudohypoxic drive (Karasek et al., 2013). Extra-adrenal chromaffin tissue normally plays an important role in catecholamine production in the developing fetus, but the tissue degenerates shortly after birth. Abnormal persistence of the fetal tissue may give rise to paraganglioma. The most common form of head and neck paraganglioma involves the hypoxia-sensing cells of the carotid body. Under hypoxic conditions, a hypoxia-inducible factor (HIF) normally translates from the cytosol to the nucleus, causing compensatory activation of the genes involved in angiogenesis, erythropoiesis, extracellular matrix turnover, and many other processes that defend against tissue hypoxia. In patients with familial paraganglioma, HIF remains activated—not by tissue hypoxia but rather by the abnormal accumulation of succinate. Such pseudohypoxic drive is also implicated in the molecular pathogenesis of tumorigenesis in VHL syndrome, as the VHL gene product normally is involved in the tonic restraint of HIF (Kaelin, 2007; Karasek et al., 2013).
Other Associated Conditions
PHEO is a great mimicker being associated with the following conditions:
- Carney Triad of secretory paraganglioma, GI stromal tumors, and pulmonary chondroma. Carney triad does not appear to be inherited, and the molecular basis for the association is unknown.
- Cholelithiasis, seen in up to 30% of PHEO patients.
- Diabetes, especially in young, lean patients (Darr et al., 2012).
- Hypercalcemia in the absence of hyperparathyroidism (Kimura et al., 1990).
- Polycythemia due to increased erythropoietin production. More frequently, a high hematocrit is related to a contracted plasma volume.
- Renovascular hypertension, likely caused by external compression of a renal artery by a paraganglioma or NE-induced vasospasm causing fibromuscular dysplasia (Sarathi et al., 2012).
- Adrenocortical hyperfunction may arise from ACTH secretion from the PHEO, a coincidental cortisol-secreting adenoma in the other adrenal, or bilateral adrenal hyperplasia (Ghander et al., 2012).
- Rhabdomyolysis, which has occurred with renal failure (Anaforoglu et al., 2008).
- Megacolon, reported in 17 cases (Sweeney et al., 2000).
Conditions Simulating PHEO
Most patients with hypertension and one or more of the manifestations of PHEO turn out not to have that diagnosis. Table 12-6 lists the many conditions that can mimic a PHEO. The most common are panic disorder and labile primary hypertension. Other common PHEO mimics are rebound hypertension from clonidine withdrawal—especially with PRN dosing—obstructive sleep apnea (Chapters 3 and 14), and baroreflex failure (Chapters 3 and 14). The latter is suggested by a remote history of the head and neck surgery with mantle field radiation therapy, recent carotid endarterectomy, or surgical excision of bilateral carotid body tumors (i.e., vagal paragangliomas).
Pseudopheochromocytoma is a vague term often used as a diagnosis of exclusion for patients with extremely labile hypertension and symptoms indistinguishable from a true PHEO but negative biochemical testing (typically on multiple occasions) (Hunt & Lin, 2008; Mann, 2008; Sharabi et al., 2007). An emotional trigger may not be apparent and yet the paroxysms can become disabling. Excessive adrenomedullary secretion of EPI has been implicated, but larger studies are needed (Hunt & Lin, 2008; Sharabi et al., 2007).
An iatrogenic form of pseudopheochromocytoma has been described in psychiatric patients treated with clozapine, a tricyclic dibenzodiazepine with complex pharmacologic actions involving α-adrenergic and serotonin receptors (Sara et al., 2013). We have seen such labile hypertension in hospitalized patients treated with third-generation antidepressant medication plus clonidine (Victor, unpublished observations, 2014).
Death from PHEO
Most deaths are related to failure to consider PHEO in patients undergoing severe stress such as nonadrenal surgery or obstetric delivery. Many deaths are unexpected and sudden; this is likely related to catecholamine-induced effects on the cardiac muscle and conduction system. Death has followed acute hemorrhagic necrosis of a PHEO after β-blocker administration. PHEO should be considered before giving a β-blocker to control thyrotoxic symptoms, as this can precipitate a PHEO crisis.
Evaluation
When and How to Screen?
The majority of hypertensive patients do not require screening for PHEO. Screening indications are listed in Table 12-8. Many are derived from the patient’s history. Screening is indicated in all patients with adrenal incidentaloma, even if the blood pressure is normal.
TABLE 12-8 When to Screen for PHEO/Paraganglioma?

A systematic approach, such as outlined in Figure 12-2, will help to avoid missing the diagnosis and to avoid unnecessary—and expensive—laboratory testing (Yu et al., 2011). There are two steps to the diagnosis: (1) biochemical determination of autonomous catecholamine hypersecretion and (2) tumor localization.
Biochemical Diagnosis
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


