As milder degrees of PA have been recognized by the wider application of the plasma aldosterone-to-renin ratio (ARR) as a screening or case-finding test, it has become increasingly clear that the majority of patients with an elevated ARR do not have autonomous hyperaldosteronism and are even less likely to have a solitary adrenal adenoma. Therefore, bilateral adrenal venous sampling (AVS), a procedure that requires considerable expertise in performance and adds considerable expense to the workup, is now recognized to be necessary for confirmation of the type of pathology (Sarlon-Bartoli et al., 2011).
The need to establish the type of pathology is critical: Adenomas usually should be surgically removed; bilateral hyperplasia should never be surgically attacked but will almost always respond to medical therapy (Colussi et al., 2013).
The clinician is left in a dilemma: As the diagnosis of PA has become easier, the recognition of the type of pathology has become more difficult. Since CT imaging and MRI are often misleading and AVS requires considerable expertise, patients increasingly need referral to a center for definitive testing, which is often difficult and always expensive.
To avoid this dilemma, this text will present the view that the ARR screening study is grossly misleading (Jansen et al, 2014), falsely labeling more than half of those who have a ratio determined, and that it should not be done except in hypertensive patients with unexplained hypokalemia, resistance to three-drug therapy, after the finding of an adrenal incidentaloma (as described in Chapter 12), or in the relatives of patients with a familial syndrome. This view does not take in all of the patients recommended for screening in the Clinical Practice Guidelines of the Endocrine Society (Funder et al., 2008) although the major categories are similar. Even if PA is sometimes missed, medical therapy—in particular the aldosterone receptor blockers spironolactone or eplerenone—will almost always control the hypertension and, if present, the hypokalemia and all of the additional harmful effects of aldosterone excess since they are mediated through the mineralocorticoid receptor. Thereby, the patients who have PA will likely be identified, while expensive laboratory procedures, invasive diagnostic tests, and unnecessary surgery will be avoided in the majority of patients who do not. This approach was initially recommended by Funder but, despite his continued eloquent and convincing argument (Funder, 2012), the remainder of the experts’ opinions have continued to insist on wide-scale, costly, and often inaccurate screening and further evaluation and surgery.
Current practice misses most patients with PA and, at the same time, mislabels many who do not have the disease. As Funder (2012) states: “we can change our mindset to include low-dose mineralocorticoid receptor antagonists as first-line treatment for newly discovered hypertensive individuals and routinely give it in the drug regimen in established hypertension…. For individuals with occult primary aldosteronism, a first-line mineralocorticoid receptor antagonist, in conjunction with a conventional antihypertensive (and perhaps a low-dose thiazide) would be game-changing. We do not have the resources to diagnose primary aldosteronism but we have the ability to treat it.”
This view, which is detailed in the remainder of this chapter, may be too abrupt to be accepted by most who have worked in this arena. However, as of now, it is the best balance between the multiple costs of diagnosis and the infrequency of the syndrome.
DEFINITIONS
PA is the syndrome resulting from the autonomous hypersecretion of aldosterone, almost always from the adrenal cortex, usually by a solitary adenoma or by bilateral hyperplasia, rarely by the variants of these two (Table 11-1). As will be described, both germline and, more commonly, somatic mutations near the selectivity filter of the potassium channel KCNJ5 have been reported (Gomez-Sanchez, 2014). The germline mutation was initially found in three members of a single family with severe hypertension and massive BAH, whereas the somatic mutations has been found only in aldosterone-producing adenomas (APAs) (Choi et al., 2011). With expanding genetic research, more such defects will likely be discovered (Williams et al, 2014).
Most hyperaldosteronism seen in clinical practice is secondary to an increase in renin–angiotensin activity in response to a reduced renal profusion as seen with renal artery stenosis or reduced intravascular volume as in chronic edematous states. The ability to measure plasma renin activity (PRA) has made the differentiation much easier, since PRA is elevated in secondary aldosteronism and suppressed in PA.
INCIDENCE
After recognition of the multiple features of aldosterone excess in a single patient who was found at surgery to have a solitary adrenal adenoma, Conn (1955) went on to characterize the syndrome. Over the next decade, Conn et al. (1965) reported a high frequency of PA, found in almost 20% of the hypertensive patients at the University of Michigan. This high prevalence was subsequently thought to reflect the nature of the patients referred to that center, highly selected and suspected of having the disease. In most series of unselected patients reported in the 1970s and 1980s, classic PA was found in fewer than 0.5% of hypertensives (Gifford, 1969; Kaplan, 1967; Sinclair et al., 1987).
However, in the early 1990s, by the use of a simple screening test—the plasma ARR—one group of investigators in Brisbane, Australia, reported the finding of PA in 8.5% of 199 patients (Gordon et al., 1993, 1994). Subsequently, an abnormal ARR has been reported in 4% to 39% of hypertensives (Kaplan, 2007), but, as will be indicated later, that alone does not establish the diagnosis. Though the incidence of PA is higher than previously thought, it is likely not to be as common as some now believe.
CLINICAL FEATURES
The disease is usually seen in patients between the ages of 30 and 50 years (though cases have been found in patients from the age of 3 to 75 years) and in women more frequently than in men. The syndrome has been recognized during pregnancy in hypokalemic patients with even higher aldosterone levels than expected and, most important, suppressed PRA (Al-Ali et al., 2007).
The classic clinical features of PA are hypertension, hypokalemia, excessive urinary potassium excretion, hypernatremia, and metabolic alkalosis (Fig. 11-1). The usual presence of these features reflects the pathophysiology of aldosterone excess.
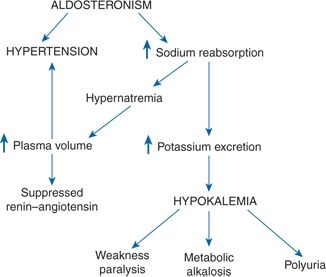
FIGURE 11-1 Pathophysiology of PA. (Reprinted from Kaplan NM. Primary aldosteronism. In: Astwood EB, Cassidy CE, eds. Clinical Endocrinology, Vol. 2. New York: Grune & Stratton; 1968:468–472, with permission.)
Hypertension
Patients with PA are hypertensive, with very few exceptions (Medeau et al., 2008). The blood pressure (BP) may be quite high—the mean in one series of 136 patients was 205/123 (Ferriss et al., 1978b). In another series of 140 patients, 28 had severe, resistant hypertension (Bravo et al., 1988), whereas in a study of 1,616 patients with resistant hypertension, PA was diagnosed in 11.3% (Douma et al., 2008). More than a dozen cases have had malignant hypertension (Kaplan, 1963; Prejbisz et al., 2013). The BP decline (dip) during the night is usually attenuated (Zelinka et al., 2004).
Looked at in another way, increased levels of aldosterone and lower levels of renin may be seen before hypertension becomes manifest. Among 3,326 normotensive participants in the Framingham Heart Study, there was a continuous gradient of increased risk of BP progression with increasing ARR levels (Newton-Cheh et al., 2007). Similar findings were noted in a 5-year follow-up of 1,984 normotensives in France (Meneton et al., 2008).
Complications
Aldosterone levels inappropriate to sodium status exert deleterious effects on various tissues by rapid, nongenomic effects through their interaction with mineralocorticoid receptors. Thereby, vascular damage (Holaj et al., 2007) and fibrosis in the heart (Diez, 2008) and kidney (Reincke et al., 2009) occur so that cardiovascular complications reflect more than the accompanying hypertension (Milliez et al., 2005; Mulatero et al, 2013a). In particular, left ventricular hypertrophy is usually disproportionate to the level and duration of hypertension (Muiesan et al., 2008).
The vascular injury is mediated, at least in part, by effects of aldosterone on the immune system (Herrada et al., 2011) that involve macrophages and T-helper effector lymphocytes, effects that can be prevented by T-regulatory lymphocytes (Kasal et al., 2012). Moreover, patients with an APA (but not those with BAH) have elevations in angiotensin II type 1 receptor autoantibodies (Kem et al, 2014; Rossitto et al., 2013).
Additional complications have been reported in patients with PA beyond the frequency seen in patients with primary (essential) hypertension, including these:
- Impairment of insulin secretion (Fischer et al., 2013), insulin resistance (Kumagai et al., 2011), and the metabolic syndrome (Vaidya et al., 2013)
- Hyperparathyroidism (Pilz et al., 2012)
- Reversible sympathetic nervous overactivity (Kontak et al., 2010)
- Anxiety (Sonino et al., 2011) and cognitive impairment (Yagi et al., 2011)
- Obesity (Rossi et al., 2011a) although they had reported that plasma aldosterone levels were correlated to body mass index in patients with primary hypertension but not in patients with PA (Rossi et al., 2008a). Even though adipocytes can produce aldosterone, the effects remain local (Briones et al., 2012).
Hemodynamics
Aldosterone infusions in conscious sheep induce hypertension by effects on the kidney (Sosa León et al., 2002), and, in humans, the hypertension is hemodynamically characterized by a slightly expanded plasma volume, an increased total body and exchangeable sodium content, and an increased peripheral resistance (Bravo, 1994; Williams et al., 1984). When 10 patients with PA, previously well controlled on spironolactone, were studied 2 weeks after the drug was stopped and the hypertension reappeared, cardiac output and sodium content (both plasma volume and total exchangeable sodium) rose initially (Wenting et al., 1982) (Fig. 11-2). Between weeks 2 and 6, the hemodynamic patterns separated into two types: In five patients, the hypertension was maintained through increased cardiac output; in the other five, cardiac output and blood volume returned to their initial values, but total peripheral resistance rose markedly. Total body sodium space remained expanded in both groups, though more so in those with increased cardiac output (Man in’t Veld et al., 1984). After surgery, the cardiac output fell in the high-flow patients, and the peripheral resistance fell in the high-resistance patients.
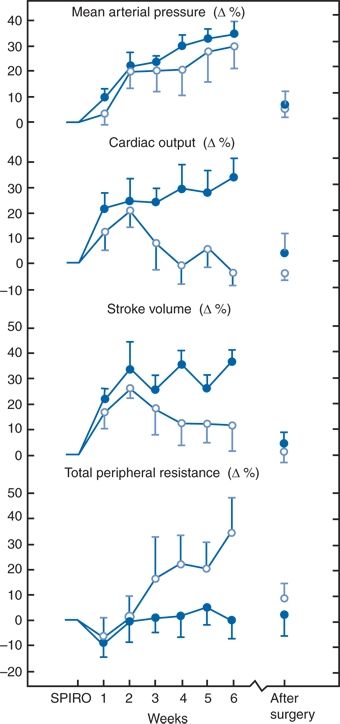
FIGURE 11-2 Changes (mean ± SEM) in systemic hemodynamics after discontinuation of spironolactone treatment (SPIRO) and after surgery in 10 patients with PA. Note the fall in stroke volume and cardiac output after 2 weeks in the five patients with high-resistance hypertension (open circles) compared to the five with high-flow hypertension (closed circles). (Reprinted from Wenting GJ, Man in’t Veld AJ, Derkx FHM, et al. Recurrence of hypertension in primary aldosteronism after discontinuation of spironolactone: Time course of changes in cardiac output and body fluid volumes. Clin Exp Hypertens 1982;A4:1727–1748, with permission.)
Mechanism of Sodium Retention
The pressor actions of aldosterone are generally related to its effects on sodium retention via its action on renal mineralocorticoid receptors (Baxter et al., 2004). Even though the kidney mineralocorticoid receptor is equally receptive to glucocorticoids and to mineralocorticoids (Arriza et al., 1987; Farman & Rafestin-Oblin, 2001), relatively small concentrations of aldosterone are able to bind to the mineralocorticoid receptor in the face of much higher concentrations of glucocorticoids (mainly cortisol) because of the action of the 11β-hydroxysteroid dehydrogenase (11β-HSD) enzyme, which converts the cortisol (with its equal affinity) into cortisone, which does not bind to the receptor (Walker, 1993).
Aldosterone stimulates sodium reabsorption through complex genomic effects that collectively act to increase the activity of the epithelial sodium channel (ENaC) in the apical membrane (Stokes, 2000). After a certain amount of persistent volume expansion, the increases in renal perfusion pressure and atrial natriuretic factor inhibit further sodium reabsorption so that “escape” from progressive sodium retention occurs, despite continued aldosterone excess (Yokota et al., 1994).
Hypokalemia
Incidence
Although normokalemia was found occasionally in the classic cases of APAs (Conn et al., 1965), hypokalemia was usual in the series reported prior to the early 1990s. In the MRC series, hypokalemia occurred in all 62 patients with a proved adenoma and was persistent in 53; among the 17 with hyperplasia, plasma potassium was persistently normal in only three patients (Ferriss et al., 1983). On the other hand, most patients in recently described series are normokalemic (Funder et al., 2008). There are a number of possible reasons why hypokalemia is now less common. These include the following:
- Most cases now being recognized are caused by BAH whose manifestations are usually milder than seen with APA. This includes the degree of potassium wastage.
- With more extensive screening, most cases are being recognized much earlier, before significant hypokalemia develops.
- Patients may experience considerable potassium loss without having the serum K+ fall to the level as defined as hypokalemia. Whereas a patient’s usual K+ level may be 4.8 mmol/L, a fall to 3.6 mmol/L may reflect significant K+ loss but not be so recognized.
- There may be a disconnect between hypertension and hypokalemia. Hypertension may develop by other nongenomic effects of aldosterone in addition to the genomic mediation of increased renal sodium reabsorption. Thereby, hypertension may develop before significant K+ wastage.
- If patients reduce the sodium intake for relief of hypertension, K+ wastage will decrease.
- Caution should be used to ensure that hypokalemia is not inadvertently missed. A number of factors may cause a temporary and spurious rise in plasma potassium, including the following: A difficult and painful venipuncture may cause plasma potassium to rise for multiple reasons: If the patient hyperventilates, the respiratory alkalosis causes potassium to leave cells; repeated fist clenching causes potassium to leave the exercising muscles; if the tourniquet is left on, plasma potassium rises from venous stasis. In a series of 152 patients with PA, serum potassium was above 3.6 mmol/L in only 10.5% in samples obtained without fist clenching but in 69.1% after fist clenching with a tourniquet in place (Abdelhamid et al., 2003).
- Any degree of hemolysis.
- Efflux of potassium from blood cells if separation of plasma by centrifugation is delayed or if the sample is placed on ice.
With significant falls in serum and body K+, aldosterone secretion may fall, even from otherwise autonomous adenomas (Kaplan, 1967). Therefore, potassium levels should be restored before aldosterone levels are measured.
Suppression of Renin Release
As a consequence of the initial expansion of vascular volume and the elevated BP, the baroreceptor mechanism in the walls of the renal afferent arterioles suppresses the secretion of renin to the point that renin mRNA may be undetectable in the kidney (Shionoiri et al., 1992). Almost all patients with PA have low levels of PRA that respond poorly to upright posture and diuretics, two maneuvers that usually raise PRA (Montori et al., 2001). Rarely, concomitant renal damage may stimulate renin release (Oelkers et al., 2000), but renin levels are almost always suppressed, even in those with malignant hypertension (Wu et al., 2000). The presence of a low renin in patients with therapy-resistant hypertension is a clue to the presence of PA (Eide et al., 2004).
Other Effects
- Hypernatremia is usual, unlike most forms of edematous secondary aldosteronism in which the sodium concentration is often quite low or with diuretic-induced hypokalemia in which slightly low serum sodium is usually found. Thus, the serum sodium concentration may provide a useful clinical separation between primary and secondary aldosteronism.
- Hypomagnesemia from excessive renal excretion of magnesium may produce tetany.
- Sodium retention and potassium wastage may be demonstrable wherever such exchange is affected by aldosterone: Sweat, saliva, and stool.
- Atrial natriuretic peptide levels are appropriately elevated for a state of volume expansion (Opocher et al., 1992).
Resistant Hypertension
Resistant hypertension refers to the persistence of BP above 140/90 mm Hg despite therapy with three antihypertensive drugs, including a diuretic, in full doses. PA has been reported to be present in 20% to 40% of patients with resistant hypertension (Calhoun, 2007) based on the findings in small groups of patients. In a larger study of 251 patients with resistant hypertension, Pimenta et al. (2007) made the diagnosis of PA in 59 patients (24%) on the basis of hormonal studies. As in other reports of a high prevalence of PA among resistant hypertensives, the patients were studied while taking a variety of drugs that can variably alter both renin and aldosterone levels. The average number of such drugs was 4.2 per patient, and 71% were on a β-blocker, which is known to lower renin more than aldosterone, giving rise to falsely positive tests for PA.
In a much more convincing study of 1,616 patients with resistant hypertension, a number far larger than the total in all previous reports, the patients were studied after all antihypertensive drugs that could alter renin and aldosterone levels were discontinued (Douma et al., 2008). PA was diagnosed in 11.3% of these patients, using multiple tests to confirm the diagnosis. The authors conclude that since resistant hypertension is found in about 10% of hypertensive patients and since PA is present in about 10% of them, the overall prevalence of PA “in the general unselected hypertensive population is much lower than currently reported.”
DIAGNOSIS
The diagnosis of PA is easy to make in patients with unprovoked hypokalemia and other manifestations of the fully expressed syndrome. The fact that hypokalemia was present in most patients in series published before 1990 likely reflects the failure to look for the syndrome in normokalemic hypertensives. Over the past decade, many more hypertensive patients have been found to have PA, the majority without hypokalemia. This higher frequency is largely the consequence of broader use of the ARR for case detection. The Clinical Practice Guideline (Funder et al., 2008) lists these groups as having a high prevalence of PA and therefore in need of testing:
- Moderate or severe hypertension, i.e., patients with systolic BP greater than 160 or diastolic BP greater than 100 mm Hg
- Resistant hypertension, defined as BP above 140/90 despite treatment with three antihypertensive medications (this definition does not require the inclusion of a diuretic)
- Hypertensives with spontaneous or diuretic-induced hypokalemia
- Hypertension with adrenal incidentaloma
The adoption of these guidelines would call for testing of a large segment of the hypertensive population, and, before their adoption, the warning by Grimes and Schulz (2002) should be noted:
Screening has a darker side that is often overlooked. It can be inconvenient, unpleasant, and expensive… A second wave of injury can arise after the initial screening insult: false-positive results and true-positive results leading to dangerous interventions.
It therefore seems prudent to restrict testing to only portions of the four groups listed in the guideline (Funder et al., 2008) for the following reasons:
- “Moderate” hypertension, from 160 to 180 systolic or 100 to 110 diastolic, would subsume about 25% of all hypertensives.
- Hypertension should not be considered “resistant” unless therapy includes a diuretic. The prevalence of apparent resistance may subsume 30% of all hypertensives although far fewer, about 10%, are truly resistant.
- Hypokalemia induced by a diuretic may reflect nothing more than an effective diuretic that induces secondary aldosteronism. If such hypokalemia is resistant to the replacement of potassium, the likelihood of PA is likely greater.
- Only about 1% of adrenal incidentalomas have been found to have PA (Young, 2007a).
Urine Potassium
Although the ARR has largely replaced other case detection testings, if hypokalemia is present, a 24-hour urine sample should be collected for sodium and potassium levels before starting potassium replacement therapy but 3 to 4 days after diuretics have been stopped. If the urine sodium is above 100 mmol/24 hours (to ensure that enough sodium is present to allow potassium wastage to express itself), the presence of a potassium level above 30 mmol/24 hours indicates a driven renal wastage of potassium. In addition to the action of excess mineralocorticoid in the syndromes of PA, a number of other conditions may require consideration, conditions in which hypokalemia is coupled with renal potassium wastage (Table 11-2).
TABLE 11-2 Causes of Hypokalemia due to Renal Loss of Potassium

Once the renal origin of hypokalemia is recognized, it may be preferable to correct the hypokalemia with potassium supplements, 40 to 80 mmol/day, after the discontinuation of diuretics before performing additional workup. To restore total body potassium deficits after a prolonged diuretic use, a minimum of 3 weeks is needed, and it may take months. After a suitable interval, the supplemental potassium should be stopped for at least 3 days and the plasma potassium level should be rechecked. If plasma potassium is normal, plasma renin and aldosterone levels should be measured. Recall that if the plasma aldosterone is not definitely elevated in the presence of hypokalemia, it should be rechecked after potassium replenishment.
Plasma Aldosterone–Renin Ratio
The ARR is derived by dividing the plasma aldosterone (normal = 5 to 20 ng/dL) by the PRA (normal = 1 to 3 ng/mL/h). If plasma aldosterone is measured in picomoles per liter and PRA in nanograms per liter, the values should be 27.7-fold higher, i.e., a ratio of 20 equals a ratio of 555 in SI units.
If plasma renin concentration (PRC)—also called “direct” or “active” renin assay—is obtained, ARR results will likely be reported as plasma aldosterone in pmol/L divided by PRC in mU/L (PA/PRC). The PRC values are approximately seven times the PRA values. In one study, PRC provided better sensitivity and specificity of the ARR than did the PRA (Lonati et al, 2014).
The ARR should be performed with attention to a number of factors that can interfere with its validity as described in the clinical practice guideline (Funder et al., 2008) (Table 11-3). Unfortunately, the number of invalidating factors keeps growing, now including the use of an antidepressant drug (Ahmed et al., 2011) or oral contraceptives (Pizzolo et al., 2010).
TABLE 11-3 Measurement of the ARR

ARR, aldosterone-to-renin ratio; PRC, plasma renin concentration; PRA, plasma renin activity. Modified from Funder JW, Carey RM, Fardella C, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: An Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2008;93:3266–3281.
The first evidence that the ARR identified more patients with PA than the small percentage previously recognized came from Gordon et al. (1993, 1994) from the Greenslopes Hospital in Brisbane, Australia. The Brisbane group found a high prevalence of an elevated ARR, 40 of 199 normokalemic hypertensives referred to their hypertension research unit. Such high prevalences have been replicated by a number of investigators in various countries throughout the world, mostly on patients referred to study centers (Table 11-4).
TABLE 11-4 The Prevalence of Autonomous Hyperaldosteronism and APAs in Patients Tested by Plasma Aldosterone to Plasma Renin Activity Ratio (ARR)a
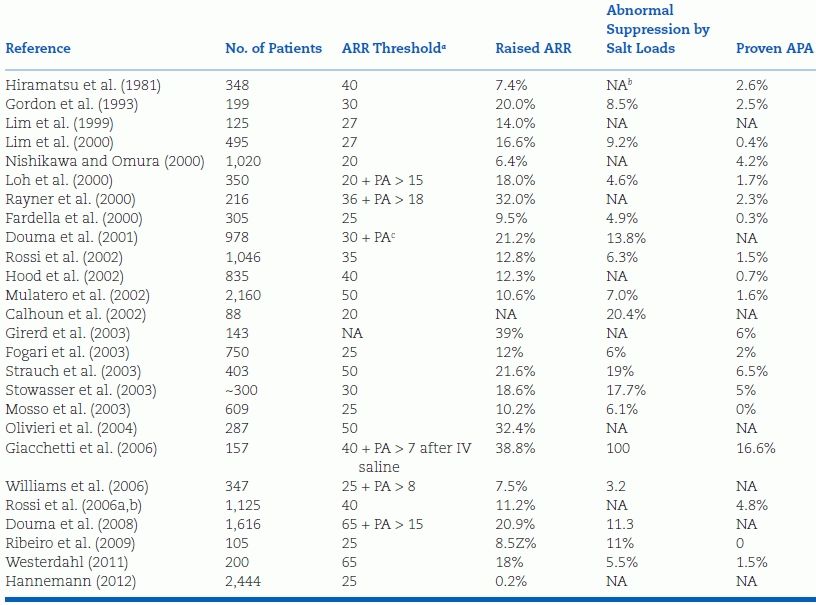
aARR expressed as plasma aldosterone in ng/dL, divided by PRA in ng/mL/h.
bNA, not available.
cincreased.
As noted in Table 11-4, there are considerable differences in the definition of an elevated ARR, with most of the ARR threshold levels representing the upper values obtained in patients presumed to have essential hypertension. The thresholds reported vary from as low as 20 to as high as 100, leading the world’s most aggressive promoter for more wide screening with the ARR to state: In essence, the ARR is a crude bivariate analysis of variables that have a skewed distribution (Rossi, 2011a,b). The reported prevalence of an elevated ARR (whatever the threshold used) in hypertensive patients varies from 6% to as high as 39% in those referred because of resistant hypertension (Kaplan, 2012). However, in by far the largest group (2,444) of truly unselected hypertensive subjects, the frequency of an elevated ARR composed of an elevated plasma aldosterone and low PRA was only 0.2% (Hannemann et al., 2012).
Despite the widespread use of the ARR to make important diagnostic and therapeutic decisions, very little study has been made of its test characteristics, i.e., sensitivity, specificity, and likelihood ratios at different cutoff values. In an attempt to better characterize the ARR, Montori et al. (2001) studied 497 patients under varying circumstances. They made two conclusions: First, the ratio varied considerably in the same patients whose posture changed from supine to standing and even more so after diuretic therapy (25 mg of hydrochlorothiazide daily) for 4 weeks; second, the ratio was “strongly and inversely dependent on the PRA level,” leading to the conclusion that “the aldosterone:renin ratio does not provide a renin-independent measure of circulating aldosterone that is suitable for determining whether plasma aldosterone concentration is elevated relative to PRA… Elevation of the ARR is predominantly an indicator of low PRA” (Montori et al., 2001).
Further concern over the sensitivity of the ARR has been raised by the data on repeated studies in 71 patients with a proven unilateral APA (Tanabe et al., 2003). The ARR was normal (below 35) in 31% of these patients on at least one occasion and only 37% had an abnormal ARR on all occasions. Rossi et al. (2010) found a much closer concordance between repeated ARR determinations.
The confounding effect of diuretic therapy noted by Montori et al. (2001) also applies to other antihypertensive medications. β-Blockers, by reducing PRA more than plasma aldosterone, can increase the number of false-positive ARRs, whereas an ACE inhibitor, angiotensin receptor blocker, or direct renin inhibitor all cause increases in PRA levels, thereby causing false-negative results (Mulatero et al., 2002). As seen in Table 11-3, the effect of these and other drugs must be considered in the preparation of the patient before the performance of the ARR test (Ahmed et al., 2011). However, in order to remove drugs that may interfere, patients may be exposed to marked rises in BP, leading to various target organ damages (Fischer et al., 2011).
As noted by Montori et al. (2001), the most common reason for false-positive ARRs is the presence of a low level of PRA as often found in the elderly, blacks, and hypertensives. With a not-unusual low PRA level of 0.3 ng/mL/h, the presence of a normal plasma aldosterone level of 12 ng/dL would provide an ARR of 40, which by most investigators’ current criteria (Table 11-4) would be abnormal.
To reduce this source of false-positive tests, some require an absolutely elevated plasma aldosterone level of 16 ng/dL or higher to call the ARR abnormal (Young, 2002). However, the Brisbane group does not, since in a description of 54 patients with documented PA, 20 had plasma aldosterone levels of 15 ng/dL or lower (Stowasser et al., 2003). Nonetheless, the wisdom of requiring an elevated plasma aldosterone has been documented, reducing false positives from 30% to 3% in one study (Seiler et al., 2004).
There is a need for establishing the “correct” cutoff of the ARR, so as to miss very few with PA, i.e., high sensitivity, and not to require additional workup in those without PA, i.e., high specificity. Many attempts have been made including a study by Bernini et al. (2008) that concluded that an ARR of 69 provided the best balance of sensitivity, 98%, and specificity, 85%. However, Jansen et al (2014) found a 5-fold difference in ARR levels obtained under the same conditions.
The continued confusion over the performance and interpretation of the ARR has led the authors of the clinical practice guideline to this conclusion:
Although it would clearly be desirable to provide firm recommendations for ARR and plasma aldosterone cutoffs, the variability of assays between laboratories and the divided literature to date make it more prudent to point out relative advantages and disadvantages, leaving clinicians the flexibility to judge for themselves. (Funder et al., 2008).
This position is clearly not suitable for the guidance of practitioners who must manage most patients. The best advice is to carefully follow all of the steps listed in Table 11-3, ensuring that patients are properly prepared and the blood sample is obtained under appropriate conditions. Then, the levels of plasma aldosterone and renin activity should be examined without calculating a ratio. If the PRA is definitely low (below 0.5 ng/mL/h) and the plasma aldosterone is definitely high (above 15 mg/dL), the same measurement of aldosterone and renin activity should be obtained on another occasion as recommended by Gordon and Stowasser (2007). If both low PRA and high aldosterone levels are found again under as careful conditions as possible, a confirmatory test should be performed.
All of this, of course, assumes that the patient is willing and able to undergo a laparoscopic adrenalectomy if the remainder of the workup confirms the presence of an APA.
Confirmatory Tests
Elevated and Nonsuppressible Aldosterone
If the PRA is low and the aldosterone is high, the presence of an inappropriately elevated and nonsuppressible aldosterone level should be documented. Four confirmatory tests have been described, with no preference given to any one of them in the 2008 guidelines. Unfortunately, the one least likely to give false-positive results, the Florinef suppression test (FST), is so difficult to perform and expensive that even its most forceful advocate states that “it is the least practical” (Stowasser, 2009).
Three of the four are based on demonstration of a lack of suppression of aldosterone after intravascular volume expansion by either intravenous or oral salt loading. This was first demonstrated with the intravenous saline suppression test of plasma aldosterone (Kem et al., 1971). Plasma aldosterone is measured before and after the infusion of 2 L of normal saline over 4 hours. Patients with PA have higher basal levels but, more importantly, fail to suppress these levels after saline to below 5 ng/dL (Mulatero et al., 2006). False-positive intravenous saline suppression tests were reported in 16.1% (Mulatero et al., 2006), 24.9% (Rossi et al., 2007), and 39% (Giacchetti et al., 2006) of patients with an elevated ARR. Therefore, to provide better specificity, a cutoff of 7 ng/dL was proposed by Giacchetti et al. (2006).
Some prefer to measure urine aldosterone levels after 3 days of oral sodium loading, with an abnormal level being above 12 (Young, 2002) or 14 μg/24 hours (Bravo, 1994). However, the Brisbane group reported that both the intravenous and oral salt loading tests are often inaccurate and they utilize a high salt diet plus large doses of the mineralocorticoid fluorohydrocortisone (Florinef) over a 4-day hospitalization, the FST test (Stowasser et al., 2003).
Captopril Suppression
Inhibition of the angiotensin-converting enzyme that converts the inactive angiotensin I to the active angiotensin II should reduce aldosterone production. Whereas plasma aldosterone levels were markedly suppressed 3 hours after oral intake of 1 mg of the ACE inhibitor captopril per kilogram of body weight in patients with primary hypertension or renovascular hypertension, they remained elevated in patients with primary hyperaldosteronism (Thibonnier et al., 1982). The normal response is a full in plasma aldosterone of 30% or more (Funder et al., 2008). Mulatero et al. (2007) reported misleading results in 4 of 11 patients. It is infrequently recommended.
Rule Out Glucocorticoid-Remediable Aldosteronism
Glucocorticoid-remediable aldosteronism (GRA) or Type I Familial Hyperaldosteronism should be considered in a young patient, particularly if other family members have aldosteronism or hemorrhagic stroke. This is most easily confirmed by demonstrating the hybrid gene in a blood sample (see below).
Excluding Other Diseases
Various causes of secondary aldosteronism are easily excluded by the presence of edema and high levels of peripheral blood PRA. In addition, there are a number of monogenic forms of hypertension, most involving renal tubular disorders and some associated with hypertension and hypokalemia, that should not be confused with PA (Stowasser & Gordon, 2006b) (Table 11-5).
TABLE 11-5 Monogenic Forms of Hypertension

Those listed under hypertension and hypokalemia also have suppressed, low PRA but all have low aldosterone levels, either because of the secretion of other mineralocorticoids (glucocorticoid-remediable hyperaldosteronism and congenital adrenal hyperplasia caused by either 11β-hydroxylase or 17- a-hydroxylase deficiency) or because of increased cortisol acting as a mineralocorticoid (apparent mineralocorticoid excess, to be covered in Chapter 13), increased sodium reabsorption from activated sodium channels (Liddle syndrome), or increased activity of mineralocorticoid receptors (Geller et al., 2000).
FAMILIAL FORMS OF HYPERALDOSTERONISM
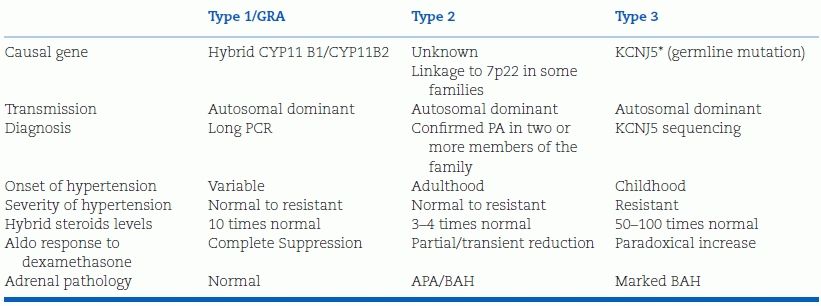
The three forms of familial hyperaldosteronism are outlined in Table 11-6.
TABLE 11-6 Clinical and Biochemical Phenotypes of Familial Forms of Hyperaldosteronism
As many as 40% of patients with sporadic, nonfamilial APA have been found to have a somatic mutation in KCNJ5 (Boulkroun et al., 2012). Unlike the very few with the germline mutation, patients with somatic mutation should be managed in the same way as those without a mutation.
PA, primary aldosteronism; APA, aldosterone-producing adenoma; BAH, bilateral adrenal hyperplasia; FH, familial hyperaldosteronism; GRA, glucocorticoid-remediable aldosteronism; KCNJ5 potassium inwardly rectifying channel, subfamily 1, member 5; CYP11B1, 11-beta–hydroxylase; CYP11B2, aldosterone synthase.
Type 1: Glucocorticoid-Remediable Aldosteronism
Sutherland et al. (1966) described a father and son with classic features of PA whose entire syndrome was completely relieved by dexamethasone, 0.5 mg four times a day (i.e., glucocorticoid remediable). Subsequently, the syndrome was shown to follow an autosomal dominant mode of inheritance and to be associated with increased levels of 18-hydroxylated cortisol. Ulick et al. (1990) postulated that the syndrome was the result of the acquisition of aldosterone synthase activity by cells of the zona fasciculata. This would explain the high levels of 18-hydroxylated steroids that can be suppressed by exogenous glucocorticoid that in turn suppresses ACTH, the normal stimulus to synthetic activity within the zona fasciculata.
Genetic Confirmation
The correctness of Ulick et al.’s postulate was proven by Lifton et al. (1992) who found “complete linkage of GRA to a gene duplication arising from unequal crossing over, fusing the 5 regulatory region of 11-beta-hydroxylase to the 3′ coding sequences of aldosterone synthase” (Fig. 11-3). The two genes lie next to one another on human chromosome 8 and are 94% identical, likely explaining the propensity to cross over (Dluhy & Lifton, 1999).
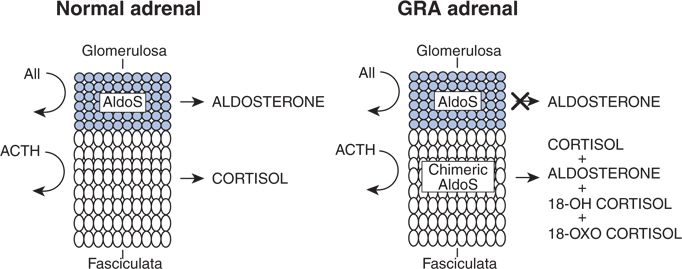
FIGURE 11-3 Regulation of aldosterone production in the zona glomerulosa and cortisol production in the zona fasciculate in the normal adrenal and model of the physiologic abnormalities in the adrenal cortex in GRA. Ectopic expression of aldosterone synthase enzymatic activity in the adrenal fasciculate results in GRA. (Reprinted from Lifton RP, Dluhy RG, Powers M, et al. Hereditary hypertension caused by chimaeric gene duplications and ectopic expression of aldosterone synthase. Nature Genet 1992;2:66–74, with permission.)
Clinical and Laboratory Features
As more patients with GRA have been identified, variations in both genotype and phenotype have been identified (Holloway et al., 2009). Different sites of gene crossover do not seem to influence the phenotype, and considerably different phenotypes have been seen within a single family that are not accounted for by different genotypes (Carajaval et al., 2012).
The hyperaldosteronism is usually evident at birth with inheritance as an autosomal dominant trait, occurring equally among men and women. The hypertension is often severe, poorly responsive to usual antihypertensive therapy, but some affected subjects in pedigrees are normotensive. An increased prevalence of strokes, particularly cerebral hemorrhage from intracranial aneurysm, has been reported (Dluhy & Lifton, 1999). About half of affected patients are normokalemic, explained by a number of factors, including a lesser mineralocorticoid activity of the 18-hydroxylated steroids and the inability of dietary potassium to stimulate aldosterone secretion when it arises from the zona fasciculata (Litchfield et al., 1997).
Diagnosis
Initially, the definitive diagnosis was based on dexamethasone suppression of aldosterone, but now that genetic testing is so readily available, this is the preferred procedure. The genetic test can be arranged by contacting the Lifton lab at Yale Medical School, either by phone at 203-737-2861 or by FAX at 203-785-3784.
Treatment
Suppressive doses of exogenous glucocorticoid will usually control the hypertension even if all the hormonal perturbations are not normalized (Stowasser et al., 2000). Spironolactone with or without a thiazide diuretic has been used without glucocorticoid suppression (Dluhy & Lifton, 1999).
Type 2: Familial Hyperaldosteronism
Familial occurrence of PA was first reported in 1991 (Gordon et al., 1991) and now has been recognized as an autosomal dominant pattern in 3-5% of members of families of patients with documented PA (Mulatero et al., 2013b), suggesting that this is by far the most common familial form. However, there are no distinguishing clinical features, there is no suppression with dexamethasone, and there is no recognized genetic defect beyond an association with linkage to a locus at chromosome 7p22 (Mulatero et al, 2013b). Therefore, these patients should be evaluated and managed in the same manner as patients with a nonfamilial type of PA.
Type 3: Familial Hyperaldosteronism
Geller et al. (2008) have reported another autosomal dominant familial form of aldosteronism with severe hypertension appearing by age 7 with very high levels of 18-hydroxylated steroids that were not suppressed by dexamethasone. Bilateral adrenalectomy was performed because of unrelenting hypertension and massive hyperplasia was found. The syndrome was later shown to be caused by a germline gain-in-function missense mutation in the KCNJ5 gene, encoding Kir3,4, a member of the inwardly rectifying K+ channel family (Choi et al., 2011). Subsequently, similar mutations were found in kindreds with milder hypertension without adrenal hyperplasia (Scholl et al., 2012) as well as in cases similar to the original with severe hypertension (Charmandari et al., 2012). Such cases are extremely rare (Pallauf et al., 2012).
Somatic Mutations in Aldosterone-Producing Adenomas
In the same paper describing the family with the germline mutation, Choi et al. (2011) also reported two somatic mutations in the same channel in 8 of 22 APAs. Subsequently, such somatic mutations have been reported in about 40% of APAs (Azizan et al., 2012; Boulkroun et al., 2012; Mulatero et al., 2013b). Such somatic mutations were not seen in hyperplastic tissue, further separating the pathophysiology of APA and BAH.
OTHER SYNDROMES
Liddle Syndrome
Liddle et al. (1963) described members of a family with hypertension, hypokalemic alkalosis, and negligible aldosterone secretion, apparently resulting from an unusual tendency of the kidneys to conserve sodium and excrete potassium even in the virtual absence of mineralocorticoids. Such patients have a mutation of the β or γ subunits of the renal ENaC, which causes increased sodium reabsorption in the distal nephron (Furuhashi et al., 2005). As will be noted in Chapter 13, these clinical features are also seen in apparent mineralocorticoid excess caused by mutations in 11β-HSD, preventing conversion of cortisol to cortisone.
Activation of Mineralocorticoid Receptor
Geller et al. (2000) identified a mutation in the mineralocorticoid receptor that causes early-onset hypertension that is markedly exacerbated in pregnancy. The exacerbation is a consequence of an altered receptor specificity so that the high levels of progesterone and other steroids lacking 21-hydroxyl groups become potent agonists.
Gordon Syndrome
In this rare syndrome, increased renal sodium and chloride retention causes hypertension and suppression of the renin–aldosterone mechanism, but with hyperkalemia (Gordon, 1986). The syndrome, known as pseudohypoaldosteronism type 2, is inherited as an autosomal dominant with at least three loci having been recognized (Disse-Nicodème et al., 2000). An elevated ARR has been noted with aldosterone stimulated by hyperkalemia and renin suppressed by volume expansion (Stowasser, 2000).
During Pregnancy
Normal pregnancy is associated with elevated plasma aldosterone but also elevated renin activity. In 31 reported cases of PA diagnosed during pregnancy, usually presenting with marked hypokalemia, renin levels were reduced (Lindsay & Nieman, 2006). Moreover, preexisting hypertension due to PA may be ameliorated during pregnancy, perhaps by antagonism of the effects of elevated aldosterone by the high progesterone levels (Murakami et al., 2000). Management is complicated by the inability to use most medical therapies, and laparoscopic adrenalectomy may be the preferred treatment.
TYPES OF ADRENAL PATHOLOGY
Once the diagnosis of PA is made, the type of adrenal pathology must be ascertained since the choice of therapy is different: Surgical for an adenoma and medical for hyperplasia. This need is even greater today than in the past as recognition of patients with milder manifestations of aldosteronism is so much easier and more frequently performed.
Aldosterone-Producing Adenomas
Solitary benign adenomas (Fig. 11-4) are almost always unilateral and most are small, weighing less than 6 g and measuring less than 3 cm in diameter. In various series, from 20% to 85% are smaller than 1 cm (Rossi et al., 2001). Histologically, most adenomas are composed of lipid-laden cells arranged in small acini or cords, similar in appearance and arrangement to the normal zona fasciculata, the middle zone of the adrenal cortex. Moreover, focal or diffuse hyperplasia, as seen in Figure 11-4, is usually present in both the remainder of the adrenal with the adenoma and the contralateral gland (Boulkroun et al., 2010).
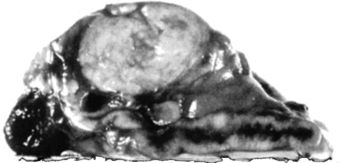
FIGURE 11-4 Solitary adrenal adenoma with diffuse hyperplasia removed from a patient with PA.
As noted, somatic mutations in genes controlling potassium channels have been reported in about 40% of APAs.
Bilateral Adrenal Hyperplasia (Idiopathic Hyperaldosteronism)
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


