10
Sensory Disorders
APPROACH TO DIAGNOSIS
An appreciation of the functional anatomy of the sensory components of the nervous system is essential for properly interpreting the history and clinical signs of patients with disorders of somatic sensation. As used here, the term includes sensations of touch or pressure, vibration, joint position, pain, temperature, and more complex functions that rely on these primary sensory modalities (eg, two-point discrimination, stereognosis, graphesthesia); it excludes special senses such as smell, vision, taste, and hearing.
FUNCTIONAL ANATOMY OF THE SOMATIC SENSORY PATHWAYS
The sensory pathway between peripheral tissues (eg, skin or joints) and the cerebral cortex involves three neurons and two central synapses (Figure 10-1).
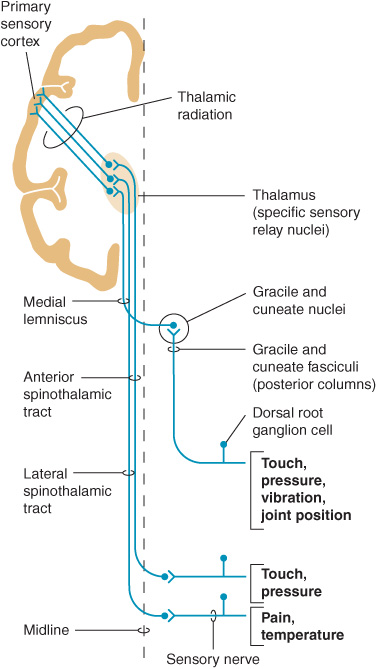
Figure 10-1. Sensory pathways conveying touch, pressure, vibration, joint position, pain, and temperature sensation. (Reproduced with permission from Barrett KE, Barman SM, Boitano S, Brooks H. Ganong’s Review of Medical Physiology. 23rd ed. New York, NY: McGraw-Hill; 2010.)
First-order sensory neurons from the limbs and trunk have cell bodies in the dorsal root ganglia. Each of these neurons sends a peripheral process that terminates in a free nerve ending or encapsulated sensory receptor and a central process that enters the spinal cord. Sensory receptors are relatively specialized for particular sensations and, in addition to free nerve endings (pain), include Meissner corpuscles, Merkel corpuscles, and hair cells (touch); Krause end-bulbs (cold); and Ruffini corpuscles (heat). First-order sensory neurons synapse centrally at a site that depends on the type of sensation. Fibers mediating touch, pressure, or postural sensation in the limbs and trunk ascend in the posterior columns of the spinal cord to the medulla, where they synapse in the gracile and cuneate nuclei. Other fibers that mediate touch and those subserving pain and temperature appreciation in the limbs and trunk synapse on neurons in the posterior horns of the spinal cord, particularly in the substantia gelatinosa. First-order sensory neurons from the face, which have cell bodies in the trigeminal (gasserian) ganglion, travel in the trigeminal (V) nerve and enter the pons. Fibers mediating facial touch and pressure synapse in the main trigeminal (V) nerve sensory nucleus, whereas those conveying facial pain and temperature synapse in the spinal trigeminal (V) nerve nucleus.
Second-order sensory neurons with cell bodies in the gracile and cuneate nuclei cross the midline and ascend in the medial lemniscus. Second-order sensory neurons that arise in the posterior horns of the spinal cord cross the mid-line and ascend in the anterolateral part of the cord: fibers mediating touch pass upward in the anterior spinothalamic tract, whereas pain and temperature fibers generally travel in the lateral spinothalamic tract. Second-order sensory neurons from the limbs and trunk are joined in the brainstem by fibers from the face: those that mediate facial touch and pressure sensation project from the main trigeminal (V) nerve sensory nucleus via the trigeminal lemniscus, and those that convey facial pain and temperature project from the spinal trigeminal (V) nerve nucleus via the trigeminothalamic tract, to the ipsilateral thalamus. In the thalamus, medial lemniscal and spinothalamic fibers synapse in the ventral posterolateral (VPL) nucleus; spinothalamic fibers also synapse in the ventral posteroinferior (VPI) nucleus and intralaminar (ILa) nuclei; and fibers in the trigeminal lemniscus and trigeminothalamic tract synapse in the ventral posteromedial (VPM) nucleus. In addition, some second-order spinothalamic sensory neurons send collaterals to the reticular formation.
Third-order sensory neurons project from the thalamus to ipsilateral cerebral cortex. Fibers from VPL, VPI, and VPM travel primarily to primary somatosensory cortex in the postcentral gyrus; fibers from ILa also project to striatum, cingulate gyrus, and prefrontal cortex.
HISTORY
Sensory disturbances may consist of loss of sensation, abnormal sensations, or pain. The term paresthesia is used to denote abnormal spontaneous sensations, such as burning, tingling, or pins and needles. The designation dysesthesia denotes any unpleasant sensation produced by a stimulus that is normally painless. The term numbness is often used by patients to describe a sense of heaviness, weakness, or deadness in the affected part of the body—and sometimes to signify any sensory impairment; the meaning must be clarified whenever this word is used.
In obtaining a history of sensory complaints, it is important to determine the location of the symptoms; the mode of onset and progression of the symptoms; whether the symptoms are constant or episodic in nature; whether any factors specifically produce, enhance, or relieve symptoms; and whether there are any accompanying symptoms.
The location of symptoms may provide a clue to their origin. For example, sensory disturbances involving all the limbs suggest peripheral neuropathy, a cervical cord or brainstem lesion, or a metabolic disturbance such as hyperventilation syndrome. Involvement of one entire limb, or of one side of the body, suggests a central (brain or spinal cord) lesion. A hemispheric or brainstem lesion may lead to lateralized sensory symptoms, but the face is also commonly affected. In addition, there may be other symptoms and signs, such as aphasia, apraxia, and visual field defects with hemispheric disease, or dysarthria, weakness, vertigo, diplopia, disequilibrium, and ataxia with brainstem disorders. Involvement of part of a limb or a discrete region of the trunk raises the possibility of a nerve or root lesion, depending on the precise distribution. With a root lesion, symptoms may show some relationship to neck or back movements, and pain is often conspicuous.
The course of sensory complaints provides a guide to their cause. Intermittent or repetitive transient symptoms may represent sensory seizures, ischemic phenomena, or metabolic disturbances such as those accompanying hyper-ventilation. Intermittent localized symptoms that occur at a consistent time may suggest the diagnosis or an exogenous precipitating factor. For example, the pain and paresthesias of carpal tunnel syndrome (median nerve compression at the wrist) characteristically occur at night and awaken the patient from sleep.
SENSORY EXAMINATION
In the investigation of sensory complaints, various modalities are tested in turn, and the distribution of any abnormality is plotted with particular reference to the normal root and peripheral nerve territories. Complete loss of touch appreciation is anesthesia, partial loss is hypesthesia, and increased sensitivity is hyperesthesia. The corresponding terms for pain appreciation are analgesia, hypalgesia, and hyperalgesia or hyperpathia; allodynia refers to the misperception of a trivial tactile sensation as pain.
PRIMARY SENSORY MODALITIES
Light Touch
The appreciation of light touch is evaluated with a wisp of cotton wool, which is brought down carefully on a small region of skin. The patient lies quietly, with the eyes closed, and makes a signal each time the stimulus is felt. The appreciation of light touch depends on fibers that traverse the posterior column of the spinal cord in the gracile (leg) and cuneate (arm) fasciculi ipsilaterally (Figures 10-1 and 10-2), passing to the medial lemniscus of the brainstem (Figure 10-3), and on fibers in the contra-lateral anterior spinothalamic tract.
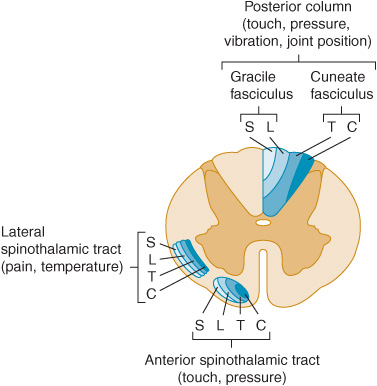
Figure 10-2. Location and lamination of sensory pathways in the spinal cord. C (cervical), T (thoracic), L (lumbar), and S (sacral) indicate the level of origin of fibers within each tract.
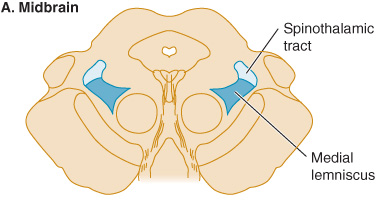
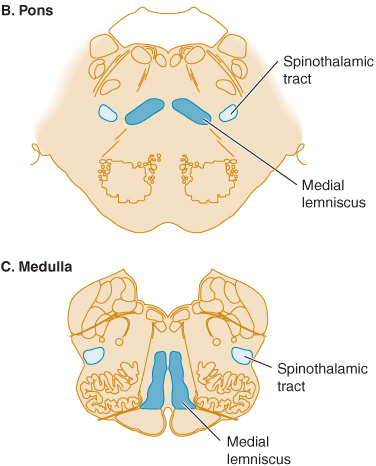
Figure 10-3. Sensory pathways in the brainstem. In the medulla, spinothalamic fibers conveying pain and temperature sensation are widely separated from medial lemniscal fibers mediating touch and pressure; these pathways converge as they ascend in the pons and midbrain.
Pinprick & Temperature
Pinprick appreciation is tested by asking the patient to indicate whether the point of a pin (not a hypodermic needle, which is likely to puncture the skin and draw blood) feels sharp or blunt. The pin should be discarded after use, and it should be handled with care because it is a potential source of infection. Appreciation of pressure or touch by the pinpoint must not be confused with the appreciation of sharpness. Temperature appreciation is evaluated by application to the skin of containers of hot or cold water. For convenience, cold can be tested by application of the side of a metal tuning fork to different bodily regions, and testing of heat is usually omitted from the routine examination. Pinprick and temperature appreciation depend on the integrity of the lateral spinothalamic tracts (Figures 10-1 and 10-2). The afferent fibers cross in front of the central canal after ascending for two or three segments from their level of entry into the cord.
Deep Pressure
Deep pressure sensibility is evaluated by pressure on the tendons, such as the Achilles tendon at the ankle.
Vibration
Vibration sense is evaluated with a tuning fork (128 Hz) that is set in motion and then placed over a bony prominence; the patient is asked to indicate whether vibration, rather than simple pressure, is felt. Many healthy elderly patients have impaired appreciation of vibration below the knees.
Joint Position
Joint position sense is tested by asking the patient to indicate the direction of small passive movements of the terminal interphalangeal joints of the fingers and toes. Patients with severe impairment of joint position sense may exhibit slow, continuous movement of the fingers (pseudoathetoid movement) when attempting to hold the hands outstretched with the eyes closed. For clinical purposes, both joint position sense and the ability to appreciate vibration are considered to depend on fibers carried in the posterior columns of the cord, although there is evidence that this is not strictly true for vibration.
COMPLEX SENSORY FUNCTIONS
Romberg Test
The patient is asked to assume a steady stance with feet together, arms outstretched, and eyes closed and is observed for any tendency to sway or fall. The test is positive (abnormal) if unsteadiness is markedly increased by eye closure—as occurs, for example, in tabes dorsalis. A positive test is indicative of grossly impaired joint position sense in the legs.
Two-Point Discrimination
The ability to distinguish simultaneous touch at two neighboring points depends on the integrity of the central and peripheral nervous system, the degree of separation of the two points, and the part of the body that is stimulated. The patient is required to indicate whether he or she is touched by one or two compass points, while the distance between the points is varied in order to determine the shortest distance at which they are recognized as different points. The threshold for two-point discrimination approximates 4 mm at the fingertips and may be several centimeters on the back. When peripheral sensory function is intact, impaired two-point discrimination suggests a disorder affecting the sensory cortex.
Graphesthesia, Stereognosis, & Barognosis
Agraphesthesia, the inability to identify a number traced on the skin of the palm of the hand despite normal cutaneous sensation, implies a lesion involving the contralateral parietal lobe. The same is true of inability to distinguish between various shapes or textures by touch (astereognosis) or impaired ability to distinguish between different weights (abarognosis).
Bilateral Sensory Discrimination
In some patients with apparently normal sensation, simultaneous stimulation of the two sides of the body reveals an apparent neglect of (or inattention to) sensation from one side, usually because of an underlying contralateral cerebral lesion.
SENSORY CHANGES & THEIR SIGNIFICANCE
It is important to determine the nature and distribution of any sensory change. Failure to find clinical evidence of sensory loss in patients with sensory symptoms must never be taken to imply that the symptoms necessarily have a psychogenic basis. Sensory symptoms often develop well before the onset of sensory signs.
PERIPHERAL NERVE LESIONS
Mononeuropathy
In patients with a lesion of a single peripheral nerve, sensory loss is usually less than would have been predicted on anatomic grounds because of overlap from adjacent nerves. Moreover, depending on the type of lesion, the fibers in a sensory nerve may be affected differently. Compressive lesions, for example, tend to affect preferentially the large fibers that subserve touch.
Polyneuropathy
In patients with polyneuropathies, sensory loss is generally symmetric and is greater distally than proximally—as suggested by the term stocking-and-glove sensory loss or length-dependent neuropathy. As a general rule, the loss will have progressed almost to the knees before the hands are affected. Sensory loss may then be accompanied by a motor deficit and reflex changes. Diabetes mellitus, amyloidosis, and certain other metabolic disorders (eg, Tangier disease, a recessive trait characterized by the near absence of high-density lipoproteins) preferentially involve small nerve fibers that subserve pain and temperature appreciation; in a pure small-fiber sensory neuropathy, the tendon reflexes are unaffected and no motor deficit occurs.
ROOT LESIONS
Nerve root involvement produces impairment of cutaneous sensation in a segmental pattern (Figure 10-4), but because of overlap there is generally no loss of sensation unless two or more adjacent roots are affected. Pain is often a conspicuous feature in patients with compressive root lesions. Depending on the level affected, there may be loss of tendon reflexes (C5-C6, biceps and brachioradialis; C7-C8, triceps; L3-L4, knee; S1, ankle), and if the anterior roots are also involved, there may be weakness and muscle atrophy.
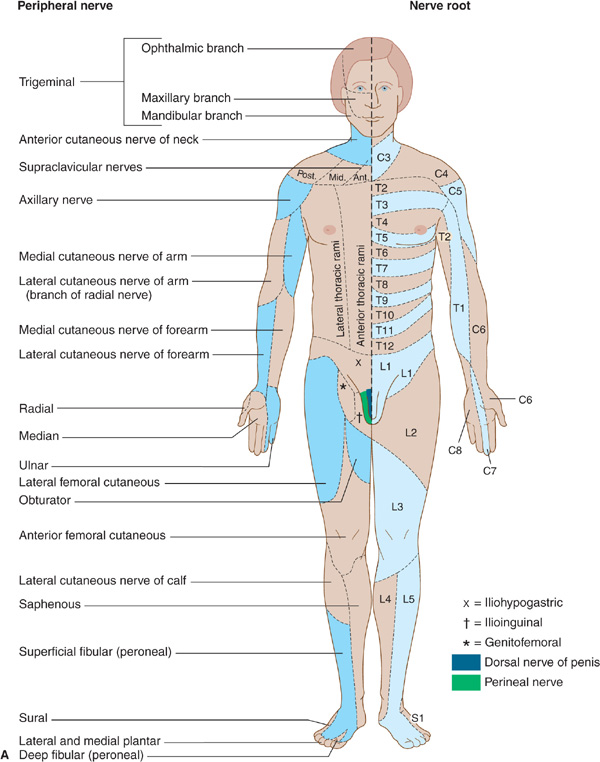
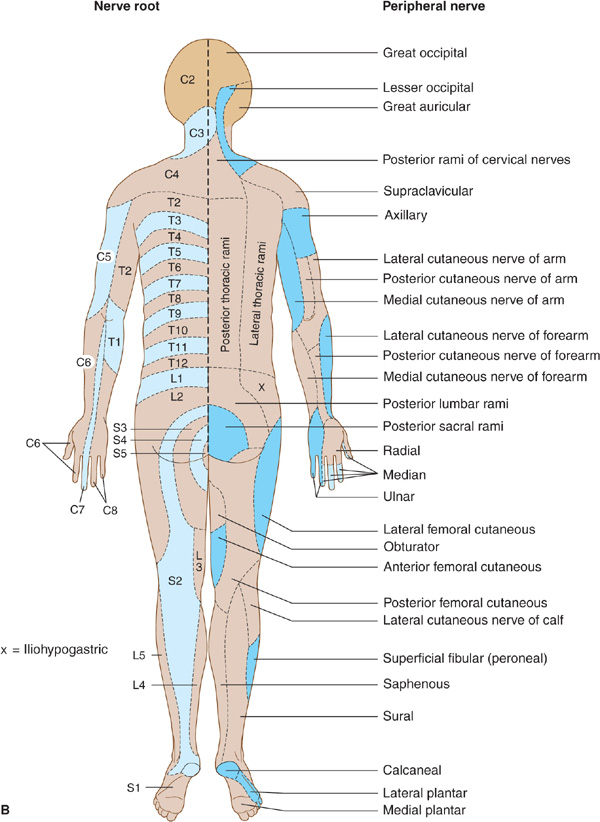
Figure 10-4. A: Cutaneous innervation (anterior view). The segmental or radicular (nerve root) distribution is shown on the left side of the body, and the peripheral nerve distribution on the right side of the body. B: Cutaneous innervation (posterior view). The segmental or radicular (nerve root) distribution is shown on the left side of the body, and the peripheral nerve distribution on the right side of the body. For details of radial, median, ulnar, fibular (peroneal), and femoral nerves, see Appendix.
CORD LESIONS
In patients with a cord lesion, there may be a transverse sensory level. Physiologic areas of increased sensitivity do occur, however, at the costal margin, over the breasts, and in the groin, and these must not be taken as abnormal. Therefore, the level of a sensory deficit affecting the trunk is best determined by careful sensory testing over the back rather than the chest and abdomen.
Central Cord Lesion
With a central cord lesion, such as occurs in syringomyelia, after trauma, and with certain cord tumors, there is characteristically a loss of pain and temperature appreciation with sparing of other modalities. This loss is due to the interruption of fibers conveying pain and temperature that cross from one side of the cord to the spinothalamic tract on the other. Such a loss is usually bilateral, may be asymmetric, and involves only the fibers of the involved segments. It may be accompanied by lower motor neuron weakness in the muscles supplied by the affected segments and sometimes by a pyramidal and posterior column deficit below the lesion (Figure 10-5).
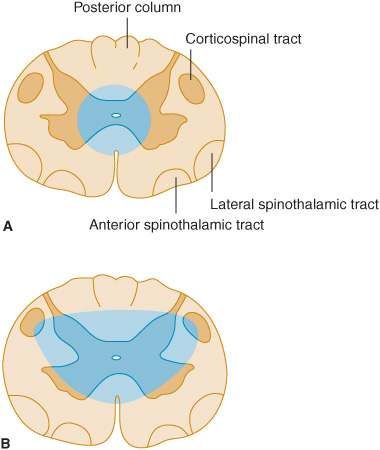
Figure 10-5. Central cord lesions (blue) of moderate (A) or marked (B) extent. Less extensive lesions impair pain and temperature appreciation by interrupting incoming sensory fibers as they cross to the contralateral spinothalamic tract; involvement of anterior horn cells causes lower motor neuron weakness. These deficits are restricted to dermatomes and muscles innervated by the involved spinal cord segments. More extensive lesions also produce disturbances of touch, pressure, vibration, and joint position sense because of involvement of the posterior columns and cause pyramidal signs because of corticospinal tract involvement, especially affecting the arms (see lamination of corticospinal tract in Figure 9-6). These deficits occur below the level of the lesion.
Anterolateral Cord Lesion
Lesions involving the anterolateral portion of the spinal cord (lateral spinothalamic tract) can cause contralateral impairment of pain and temperature appreciation in segments below the level of the lesion. The spinothalamic tract is laminated, with fibers from the sacral segments the outermost. Intrinsic cord (intramedullary) lesions often spare the sacral fibers, whereas extramedullary lesions, which compress the cord, tend to involve these fibers as well as those arising from more rostral levels.
Anterior Cord Lesion
With destructive lesions involving predominantly the anterior portion of the spinal cord, pain and temperature appreciation are impaired below the level of the lesion from lateral spinothalamic tract involvement. In addition, weakness or paralysis of muscles supplied by the involved segments of the cord results from damage to motor neurons in the anterior horn. With more extensive disease, involvement of the corticospinal tracts in the lateral funiculi may cause a pyramidal deficit below the lesion. There is relative preservation of posterior column function (Figure 10-6). Ischemic myelopathies caused by occlusion of the anterior spinal artery take the form of anterior cord lesions.
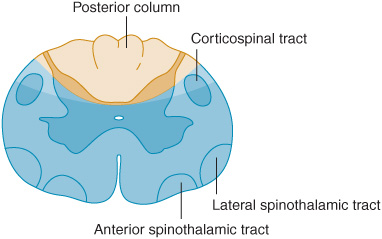
Figure 10-6. Anterior cord lesion (blue) associated with occlusion of the anterior spinal artery. Clinical features are similar to those seen with severe central cord lesions (Figure 10-5B), except that posterior column sensory functions are spared and the defect in pain and temperature sensation extends to sacral levels.
Posterior Column Lesion
A patient with a posterior column lesion may complain of a tight or bandlike sensation in the regions corresponding to the level of spinal involvement and sometimes also of paresthesias (like electric shocks) radiating down the extremities on neck flexion (Lhermitte sign). There is loss of vibration and joint position sense below the level of the lesion, with preservation of other sensory modalities. The deficit may resemble that resulting from involvement of large fibers in the posterior roots.
Cord Hemisection
Lateral hemisection of the cord leads to Brown-Séquard syndrome. Below the lesion, there is an ipsilateral pyramidal deficit and disturbed appreciation of vibration and joint position sense, with contralateral loss of pain and temperature appreciation that begins two or three segments below the lesion (Figure 10-7). Hyperalgesia and spontaneous pain are sometimes prominent ipsilaterally.
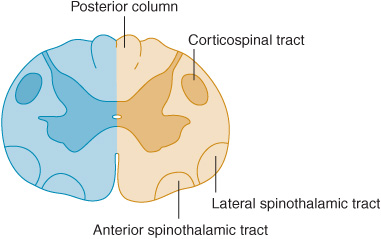
Figure 10-7. Cord lesion (blue) in Brown-Séquard syndrome. Hemisection of the cord causes ipsilateral pyramidal dysfunction and impairment of posterior column sensory function below the level of the lesion, and contralateral impairment of pain and temperature sensation with an upper limit slightly below the level of the lesion.
BRAINSTEM LESIONS
Sensory disturbances may be accompanied by a motor deficit, cerebellar signs, and cranial nerve palsies when the lesion is in the brainstem.
In patients with lesions involving the spinothalamic tract in the dorsolateral medulla and pons, pain and temperature appreciation are lost in the limbs and trunk on the opposite side of the body. When such a lesion is located in the medulla, it also typically involves the spinal trigeminal nucleus, impairing pain and temperature sensation on the same side of the face as the lesion. The result is a crossed sensory deficit that affects the ipsilateral face and contralateral limbs. In contrast, spinothalamic lesions above the spinal trigeminal nucleus affect the face, limbs, and trunk contralateral to the lesion. With lesions affecting the medial lemniscus, there is loss of touch and proprioception on the opposite side of the body. In the upper brainstem, the spinothalamic tract and medial lemniscus run together so that a single lesion may cause loss of all superficial and deep sensation over the contralateral side of the body (Figure 10-3).
THALAMIC LESIONS
Thalamic lesions may lead to loss or impairment of all forms of sensation on the contralateral side of the body, and this may have a distribution that differs from the area of symptomatic involvement. Spontaneous pain, sometimes with a particularly unpleasant quality, may occur on the affected side. Patients may describe it as burning, tearing, knifelike, or stabbing, but often have difficulty characterizing it. Any form of cutaneous stimulation can lead to painful or unpleasant sensations. Such a thalamic syndrome (Dejerine-Roussy syndrome) can also occasionally result from lesions of the white matter of the parietal lobe or from cord lesions, as discussed later.
LESIONS OF THE SENSORY CORTEX
Disease limited to the sensory cortex impairs discriminative sensory function on the opposite side of the body. Thus patients may be unable to localize stimuli on the affected side or to recognize the position of different parts of the body. They may not be able to recognize objects by touch or to estimate their size, weight, consistency, or texture. Cortical sensory disturbances are usually more conspicuous in the hands than in the trunk or proximal portions of the limbs.
DISTINCTION BETWEEN ORGANIC & PSYCHOGENIC SENSORY DISTURBANCES
Psychogenic disturbances of sensation may be associated with such psychiatric disturbances as conversion disorder. They may take any form but most often are restricted to loss of cutaneous sensation. There may be several characteristic features.
Nonorganic sensory loss does not conform in its distribution to any specific neuroanatomic pattern. It may surround a bony landmark or involve an area defined by surface landmarks rather than innervation. Indeed, it is not uncommon for there to be an apparent loss of sensation in one or more extremities, with the margin occurring circumferentially in the axilla or groin; organic sensory loss with such a margin is unusual. Organic peripheral sensory loss over the trunk or face does not usually extend to the midline but stops 3 to 5 cm before it, because of overlap in the innervation on the two sides; with nonorganic disturbances, apparent sensory loss commonly stops precisely at the midline.
There is often a sudden transition between areas of nonorganic sensory loss and areas with normal sensation. By contrast, with organic disturbances, there is usually an area of altered sensation between insensitive areas and adjacent areas with normal sensibility.
In nonorganic disturbances, there may be a dissociated loss that is difficult to interpret on an anatomic basis. For example, there may be a total loss of pinprick appreciation but preserved temperature sensation. Moreover, despite the apparent loss of posterior column function, the patient may be able to walk normally or maintain the arms outstretched without difficulty or pseudoathetoid movements.
In nonorganic sensory disturbances, appreciation of vibration may be impaired on one side but not the other side of a bony midline structure, such as the skull or sternum. The vibrations are in fact conducted to both sides by the bone, so that even if there is a hemisensory disturbance, the vibrations are appreciated on either side in patients with organic sensory disorders.
Finally, it should be noted that sensory disturbances are often suggested to the patient by the examiner’s own expectations. Such findings can be particularly misleading because they may be neuroanatomically correct. One helpful approach is to have the patient outline on the body the extent of any perceived sensory disturbance before formal sensory testing is undertaken.
PERIPHERAL NERVE LESIONS
Sensory symptoms are usually a conspicuous feature in patients with peripheral nerve lesions (Table 10-1). Sensory impairment may be in a distal stocking-and-glove pattern in patients with polyneuropathies or may follow the pattern of individual peripheral nerves in patients with mononeuropathies (Figure 10-4).


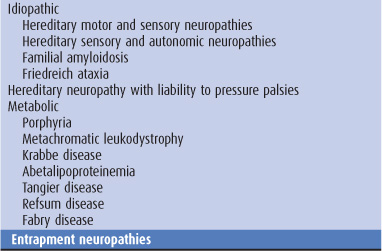
Table 10-1. Causes of peripheral neuropathy.
CLASSIFICATION
MONONEUROPATHY SIMPLEX
This term signifies involvement of a single peripheral nerve.
MONONEUROPATHY MULTIPLEX
In this disorder, several individual nerves are affected, usually at random and noncontiguously. Clinical examination reveals a clinical deficit attributable to involvement of one or more isolated peripheral nerves, except when mononeuropathy multiplex is extensive and the resulting deficits become confluent.
POLYNEUROPATHY
The term polyneuropathy denotes a disorder in which the function of numerous peripheral nerves is affected at the same time. This leads to a predominantly distal and symmetric deficit, with loss of tendon reflexes except when small fibers are selectively involved. Polyneuropathies are sometimes subclassified according to the primary site at which the nerve is affected.
In distal axonopathies (axonal neuropathies), the axon is the principal pathologic target; most polyneuropathies fall into this category.
Myelinopathies (demyelinating neuropathies) are conditions that involve the myelin sheath surrounding the axon. These disorders include acute idiopathic polyneuropathy (Guillain-Barré syndrome), chronic inflammatory demyelinating neuropathy, diphtheria, certain paraneoplastic and paraproteinemic states, and various hereditary conditions including metachromatic leukodystrophy, Krabbe disease, and types 1 and 3 Charcot-Marie-Tooth hereditary motor and sensory neuropathy (CMT-1 and 3).
CLINICAL FINDINGS
SENSORY DISTURBANCES
Involvement of sensory fibers can lead to numbness and impaired sensation; abnormal spontaneous sensations, such as pain and paresthesias; and perverted sensations such as hyperpathia (amplified response to a normally painful stimulus).
Pain
Pain is a conspicuous feature of certain neuropathies, especially if small fibers within the nerves are affected. The precise mechanism of its genesis is unclear. Polyneuropathies associated with prominent pain include those related to diabetes, alcoholism, porphyria, Fabry disease, amyloidosis, rheumatoid arthritis, and acquired immunodeficiency syndrome (AIDS), as well as dominantly inherited sensory neuropathy and paraneoplastic sensory neuronopathy. Pain is also a feature of many entrapment neuropathies and of idiopathic brachial plexopathy.
Dissociated Sensory Loss
Dissociated sensory loss is impairment of some sensory modalities, such as pain and temperature, with preservation of others, such as light touch, vibration, and joint position sense. Although the presence of a dissociated sensory loss often indicates a spinal cord lesion, it also occurs in peripheral neuropathies when there is selective involvement of peripheral nerve fibers of a certain size, such as occurs in amyloid neuropathy, leprous neuritis, or hereditary sensory neuropathy. Small fiber disease is commonly associated with disproportionate impairment of pain and temperature appreciation, spontaneous pain, and autonomic dysfunction. Large fiber disease, by contrast, results in defective touch, vibration, and joint position sense, early loss of tendon reflexes, and prominent motor symptoms.
MOTOR DEFICITS
The motor deficit that occurs with a peripheral nerve lesion consists of weakness of muscles innervated by the nerve, accompanied in severe cases by wasting and fasciculation. There may be difficulty in the performance of fine tasks; this is compounded by any accompanying sensory loss. The clinical findings reflect a lower motor neuron deficit, and it is the distribution of these signs and the presence of accompanying sensory and reflex changes that suggest they may be due to peripheral nerve involvement.
TENDON REFLEXES
These are impaired or lost if reflex arcs are interrupted on either the afferent or efferent side (C5-C6, biceps and brachioradialis; C7-C8, triceps; L3-L4, knee; S1, ankle). The ankle reflexes are usually the first to be lost in patients with polyneuropathies, but may also be absent in healthy elderly subjects.
AUTONOMIC DISTURBANCES
Autonomic disturbances may be particularly conspicuous in some peripheral neuropathies—especially Guillain-Barré syndrome and neuropathies related to diabetes, renal failure, porphyria, certain paraneoplastic disorders, or amyloidosis. Symptoms include postural hypotension, coldness of the extremities, impaired thermoregulatory sweating, disturbances of bladder and bowel function, and impotence.
ENLARGED NERVES
Palpably enlarged peripheral nerves raise the possibility of leprosy, amyloidosis, hereditary motor and sensory neuropathies, Refsum disease, acromegaly, or chronic inflammatory demyelinating polyneuropathy.
EVALUATION OF PATIENTS
TIME COURSE
Polyneuropathy that develops acutely over a few days usually relates to an inflammatory process, as in the Guillain-Barré syndrome. It may also relate to an underlying neoplasm, to infections such as diphtheria, to metabolic disorders such as acute intermittent porphyria, or to exposure to such toxic substances as thallium or triorthocresyl phosphate. A chronic course with a gradual evolution over several years is typical of many hereditary or metabolic polyneuropathies but also characterizes chronic inflammatory demyelinating polyneuropathy.
Mononeuropathy of acute onset is likely to be traumatic or ischemic in origin, whereas one evolving gradually is more likely to relate to entrapment (ie, compression by neighboring anatomic structures) or to recurrent minor trauma.
AGE AT ONSET
Polyneuropathy that develops during childhood or early adult life often has a hereditary basis, but may also relate to an underlying inflammatory disorder. Polyneuropathy developing in later life is more likely to result from a metabolic, toxic, or inflammatory disorder, or from an underlying neoplasm.
Mononeuropathy presenting in the neonatal period is likely to be developmental in origin or related to birth injury; one developing in later life may relate to entrapment or injury that is often occupationally determined.
OCCUPATIONAL HISTORY
Various industrial toxins can lead to peripheral neuropathy, including carbon disulfide, n-hexane, ethylene oxide, methyl bromide, acrylamide, triorthocresyl phosphate and certain other organophosphates, DDT, arsenic, lead, and thallium. A mononeuropathy is sometimes the first clinical manifestation of an occupationally related polyneuropathy, but it may also develop in response to entrapment or recurrent minor occupational trauma. For example, carpal tunnel syndrome is more common in persons who do heavy manual labor or develop repetitive motion injury as a result of computer terminal use, and a lesion of the deep palmar branch of the ulnar nerve may relate to repeated pressure on the palm of the hand by, for example, punching down heavily on a stapler or using heavy equipment such as a pneumatic road drill.
MEDICAL HISTORY
Metabolic Disorders
Peripheral neuropathy may relate to metabolic disorders such as diabetes mellitus, uremia, liver disease, myxedema, acromegaly, metachromatic leukodystrophy, or Fabry disease. That caused by diabetes is especially important and may take the form of an entrapment mononeuropathy, acute ischemic mononeuropathy, distal sensorimotor polyneuropathy, subacute proximal motor polyradiculopathy (diabetic amyotrophy), thoracoabdominal radiculopathy, or autonomic neuropathy.
Neoplasm
A peripheral neuropathy may also relate to an underlying malignant neoplasm. The peripheral nerves, spinal nerves, and limb plexuses may be compressed or infiltrated by extension of primary tumors or metastatic lymph nodes. Neoplastic disease can also lead to a nonmetastatic (paraneoplastic) sensory or sensorimotor polyneuropathy or to Lambert-Eaton syndrome, a disorder of neuromuscular transmission discussed in Chapter 9.
Connective Tissue Disorders
Certain connective tissue disorders, especially polyarteritis nodosa, rheumatoid arthritis, Churg-Strauss syndrome, and Wegener granulomatosis, may be associated with mononeuropathy multiplex or, less commonly, polyneuropathy, or cranial neuropathy. Polyneuropathy is more common in systemic lupus erythematosus. Patients with rheumatoid arthritis are particularly likely to develop focal entrapment or compressive mononeuropathies in the vicinity of the affected joints.
AIDS
Acquired immune deficiency syndrome (AIDS) is commonly associated with a distal, symmetric, primarily sensory polyneuropathy. Peripheral nerve involvement in AIDS less frequently takes the form of an acute or chronic inflammatory demyelinating polyneuropathy, polyradiculopathy, mononeuropathy multiplex, or autonomic neuropathy. Neuropathies are also seen in patients with asymptomatic human immunodeficiency virus-1 (HIV-1) infection and HIV-1 seroconversion.
DRUG & ALCOHOL HISTORY
Some of the drugs that cause peripheral neuropathy are listed in Table 10-2; there may be selective involvement of motor or sensory fibers with some drugs.

Table 10-2. Selected drugs inducing peripheral neuropathy.
FAMILY HISTORY
Certain polyneuropathies have a hereditary basis. These are discussed later in this chapter in the section on hereditary neuropathies.
DIFFERENTIAL DIAGNOSIS
Peripheral neuropathies can lead to a motor or sensory deficit or both. The preservation of sensation and tendon reflexes distinguishes the motor deficit that results from pure pyramidal lesions or is associated with spinal muscular atrophies, myopathies, or disorders of neuromuscular transmission from that caused by peripheral nerve involvement. Other distinguishing features are discussed in Chapter 9.
Myelopathies are characterized by a pyramidal deficit below the level of the lesion as well as by distal sensory loss. In tabes dorsalis, there is often a history of syphilitic infection, and examination reveals other stigmas of syphilis. In addition, tactile sensation is preserved.
Radiculopathies are distinguished from peripheral neuropathies by the distribution of motor or sensory deficits (Figure 10-4). The presence of neck or back pain that radiates to the extremities in a radicular distribution also suggests a root lesion.
INVESTIGATIVE STUDIES
Laboratory studies in patients with peripheral neuropathy are directed at confirming the diagnosis and revealing any underlying cause.
Electromyography may reveal evidence of denervation in the affected muscles and can be used to determine whether any motor units remain under voluntary control. Nerve conduction studies permit conduction velocity to be measured in motor and sensory fibers. On the basis of electro-diagnostic or histopathologic studies, peripheral neuropathies can be divided into demyelinating or axonal neuropathies. In demyelinating neuropathies, electro-myography typically reveals little or no evidence of denervation, but there is conduction block or marked slowing of maximal conduction velocity in affected nerves. In axonal neuropathies, electromyography shows that denervation has occurred, especially distally in the extremities, but maximal nerve conduction velocity is normal or slowed only slightly.
Laboratory studies that may help to identify the cause of a peripheral nerve disorder include a complete blood count, erythrocyte sedimentation rate, serum urea nitrogen and creatinine, fasting blood glucose, serum vitamin B12, serum protein, protein electrophoresis and immuno-electrophoresis, liver and thyroid function blood tests, serologic test for syphilis (FTA or MHA-TP), rheumatoid factor, antinuclear antibody, and chest x-ray. Depending on the clinical circumstances, serologic tests for Lyme disease, hepatitis, infection with HIV, or paraneoplastic antibodies may be required. Genetic studies may also be necessary after appropriate genetic counseling.
If toxic causes are suspected, a 24-hour urine collection followed by analysis for heavy metals may be necessary, and hair and fingernail clippings can be analyzed for arsenic. Examination of a fresh specimen of urine for porphobilinogen and δ-aminolevulinic acid is necessary if porphyria is suspected.
TREATMENT
DISEASE-SPECIFIC TREATMENT
Treatment of the underlying cause may limit the progression of or even reverse the neuropathy. Disease-specific treatments are discussed later under individual disorders.
VENTILATORY ASSISTANCE
Respiratory function must be monitored carefully—particularly in acute idiopathic polyneuropathy (Guillain-Barré syndrome), chronic inflammatory demyelinating polyneuropathy, and diphtheritic neuropathy—and preparations must be made to assist ventilation if the forced vital capacity reaches 15 mL/kg, the mean inspiratory force reaches –40 mm Hg, dyspnea becomes evident, or the oxygen saturation of arterial blood declines.
TRAUMA PREVENTION
Nursing care is important in patients with severe motor or sensory deficits to prevent decubitus ulcers, joint contractures, and additional compressive peripheral nerve damage. In patients with severe dysesthesia, a cradle (inverted metal bar frame) can be used to keep the bedclothes from touching sensitive areas of the skin.
Extremities with sensory loss must be protected from repeated minor trauma, such as thermal injury, that can destroy tissues. The temperature of hot surfaces should be checked with a part of the body in which sensation is preserved, and the setting of water heaters must be reduced to prevent scalding. The skin and nails must be cared for meticulously.
PAIN RELIEF
Patients with sensory disorders may suffer from especially severe pain, which can dramatically impair quality of life. Treatment of this pain should be a high priority.
Duloxetine (60 mg once daily) or venlafaxine (titrated up to 75 mg two to three times daily in standard formulation or given as the equivalent dose once daily in the extended release formulation) is often helpful; both are selective serotonin and norepinephrine reuptake inhibitors.
Pregabalin (150 mg increasing after 1 week to 300 mg daily in divided doses; maximum, 600 mg daily) is also particularly useful for the relief of neuropathic pain.
Phenytoin 300 mg/d, carbamazepine up to 1,200 mg/d, or mexiletine 600 to 900 mg/d can sometimes relieve the lancinating pain of certain neuropathies. If the pain is more constant, burning, or dysesthetic, amitriptyline 25 to 100 mg at bedtime is often helpful as are other tricyclic agents.
Topical capsaicin is also helpful in neuropathic pain syndromes.
AUTONOMIC DISTURBANCES
Dysautonomic symptoms may be troublesome in several polyneuropathies, but especially in diabetic polyneuropathy. Waist-high elastic hosiery, dietary salt supplementation, and treatment with fludrocortisone 0.1 to 1 mg/d orally may help relieve postural hypotension, but the patient must be monitored carefully to prevent recumbent hypertension. Other medications that may be helpful include clonidine, midodrine, dihydroergotamine, octreotide, or beta-blockers. Instructing the patients to sleep in a semierect rather than a recumbent position is helpful because dysautonomic patients are often unable to conserve salt and water when recumbent at night.
POLYNEUROPATHIES
IDIOPATHIC INFLAMMATORY NEUROPATHIES
ACUTE IDIOPATHIC POLYNEUROPATHY (GUILLAIN-BARRÉ SYNDROME)
Guillain-Barré syndrome is an acute or subacute polyneuropathy that can follow minor infective illnesses, inoculations, or surgical procedures or may occur without obvious precipitants. Clinical and epidemiologic evidence suggests an association with preceding Campylobacter jejuni infection. Its precise cause is unclear, but it appears to have an immunologic basis. Both demyelinating and axonal forms have been recognized, with distinctive clinical and electro-physiologic features. The demyelinative form is more common in the United States, but an axonal variant is encountered occasionally (acute motor sensory axonal neuropathy). In northern China, a related axonal form occurs frequently and has been designated acute motor axonal neuropathy.
Clinical Features
The features useful for diagnosing Guillain-Barré syndrome are summarized in Table 10-3. Patients generally present with ascending weakness that is symmetric, usually begins in the legs, is often more marked proximally than distally, and is sometimes so severe that it is life-threatening, especially if the muscles of respiration or swallowing are involved. Muscle wasting develops if axonal degeneration has occurred. Sensory complaints, although usually less marked than motor symptoms, are also frequent. The deep tendon reflexes are typically absent. There may be marked autonomic dysfunction, with tachycardia, cardiac irregularities, labile blood pressure, disturbed sweating, impaired pulmonary function, sphincter disturbances, paralytic ileus, and other abnormalities.
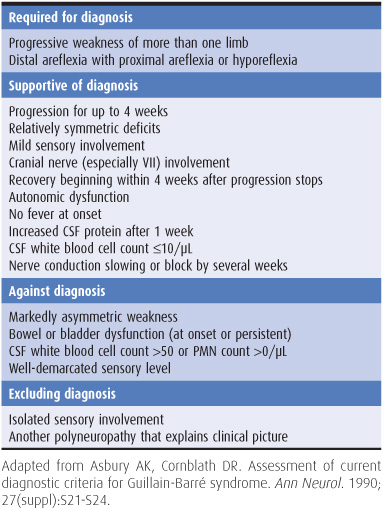
Table 10-3. Diagnostic criteria for Guillain-Barré syndrome.
Investigative Studies
The cerebrospinal fluid (CSF) often shows a characteristic abnormality, termed cytoalbuminologic dissociation, with increased protein concentration but a normal cell count; abnormalities may not be found in the first week, however. Electrophysiologic studies may reveal marked slowing of motor and sensory conduction velocity or evidence of denervation and axonal loss. The time course of the electrophysiologic changes does not necessarily parallel any clinical developments. When HIV-1 infection is suspected because of the clinical context in which the neuropathy has developed or the presence of high-risk factors, appropriate serologic studies should be performed.
Treatment
Plasmapheresis appears to reduce the time required for recovery and may decrease the likelihood of residual neurologic deficits. It is best instituted early, and it is indicated especially in patients with a severe or rapidly progressive deficit or respiratory compromise. Intravenous immunoglobulin (400 mg/kg/d for 5 days) appears to be equally effective and should be used in preference to plasmapheresis in adults with cardiovascular instability and in children; the two therapies are not additive.
Therapy is otherwise symptomatic, the aim being to prevent such complications as respiratory failure or vascular collapse. For this reason, patients who are severely affected are best managed in intensive care units, where facilities are available for monitoring and assisted respiration if necessary (eg, if the forced vital capacity reaches 15 mL/kg, the mean inspiratory force reaches –40 mm Hg, the patient is short of breath, or the blood oxygen saturation declines). Volume replacement or treatment with pressor agents is sometimes required to counter hypotension, and low-dose heparin may help to prevent pulmonary embolism. Corticosteroids may affect the outcome adversely or delay recovery, and are not indicated.
Prognosis
Symptoms and signs cease to progress by approximately 4 weeks into the illness. The disorder is self-limiting, and improvement occurs over the weeks or months after onset. Approximately 70% of patients recover completely, 25% are left with mild neurologic deficits, and 5% die, usually as a result of respiratory failure. The prognosis is poorer when there is evidence of preceding Campylobacter jejuni infection, and a more protracted course and less complete recovery are also likely when axonal degeneration rather than demyelination is the primary pathology. Advanced age, the need for ventilatory support, or more rapid onset of symptoms may also predict a poorer prognosis.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


