It is a myopathy, as defined by clinical, histologic, and electromyographic (EMG) criteria. No signs of denervation or sensory loss are apparent unless there is a concomitant and separate disease.
All symptoms are effects of limb or cranial muscle weakness. (The heart and visceral muscles may also be involved.)
Symptoms become progressively worse.
Histologic changes imply degeneration and regeneration of muscle, but no abnormal storage of a metabolic product is evident.
The condition is recognized as heritable, even if there are no other cases in a particular family.
It is incompletely understood why muscles are weak in any of these conditions, even when the affected gene product is known.
There is no curative therapy for any of the dystrophies yet, and gene therapy has not yet been successful.
Muscle ultrasound is painless, less expensive compared to MRI, and offers dynamic images. Muscle imaging can show the selective pattern of muscle involvement associated with some of the muscular dystrophies. For example, there is selective involvement of sartorius and adductor longus in selenoprotein N1 (SEPN1) myopathy with sparing of the gracilis. MRI also offers good delineation between fatty infiltration and skeletal muscle and may guide the choice of the muscle to biopsy.
TABLE 142.1 Features of the Most Common Muscular Dystrophies | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
cases are caused by de novo mutations; the others are more clearly familial. Because the life span of patients with DMD is shortened, the prevalence is less—about 1 in 18,000 males. Becker muscular dystrophy (BMD) is much less common, with a frequency of about 1 in 20,000.
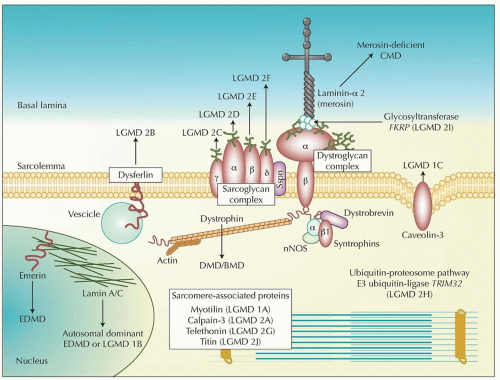 FIGURE 142.1 Skeletal muscle proteins and muscular dystrophies. The dystrophin-glycoprotein complex (DGC) comprises dystrophin, the dystroglycans (α, β), the sarcoglycans (α, β, γ, δ), sarcospan, the syntrophins (α, β1), and dystrobrevin (α). The complex stabilizes the sarcolemma and protects surface membranes in muscle contraction. Nitric oxide synthetase (nNOS) interacts with the syntrophin complex and laminin-α 2, one of many extracellular ligands of α-dystroglycan. Disease-causing mutations are cited in the text and Table 142.3. LGMD, limb-girdle muscular dystrophy; CMD, congenital muscular dystrophy; DMD, Duchenne muscular dystrophy; BMD, Becker muscular dystrophy; EDMD, Emery-Dreifuss muscular dystrophy. (From Mathews KD, Moore SA. Limb-girdle muscular dystrophy. Curr Neurol Neurosci Rep. 2003;3:78-85, with permission.) |
ocular movements are spared. Iliotibial contractures limit hip extension; heel cord contractures are partly responsible for toe walking. Loss of ambulation occurs between ages 9 and 12 years or later when treated with steroids. Boys then become wheelchairbound. Scoliosis may then become serious and may compromise limb and respiratory function. Elbow and knee contractures contribute to disability. Respiratory muscle weakness causes progressive decline in lung capacity beginning at about age 8 years. Nocturnal hypoventilation may occur and cause morning headaches and daytime fatigue if untreated. By about age 20 years, respiration is severely compromised that respiratory support is needed. Life expectancy in DMD has improved greatly in the last three decades, probably because of better coordinated medical care and home nocturnal ventilation. Some attribute the improvement to prednisone therapy.
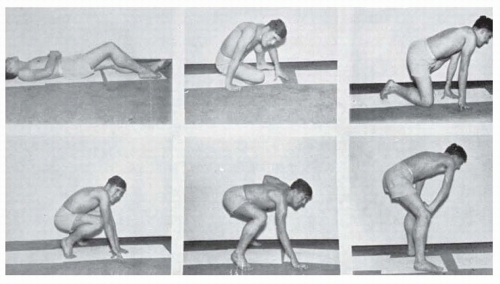 FIGURE 142.2 Gowers sign in a patient with DMD or BMD. Postures assumed in attempting to rise from the supine position. |
asymptomatic individuals (idiopathic or asymptomatic hyperCK-emia). Even before myopathic symptoms begin, high CK values are seen in the dysferlinopathies (LGMD2B or Miyoshi myopathy), which are discussed later in the chapter. Similarly, caveolin-3 mutations (LGMD 1C) may cause hyperCKemia before symptoms of limb-girdle dystrophy become evident.
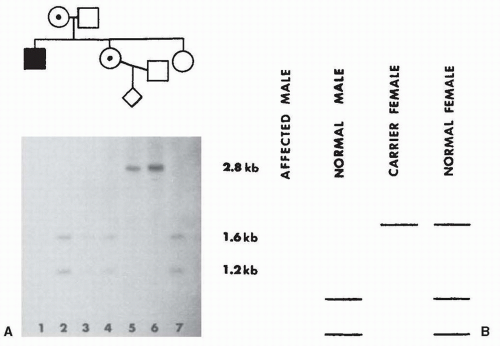 FIGURE 142.3 Prenatal diagnosis in DMD. A: Autoradiograph of Southern blot of 1% agarose gel with DNA samples from each individual digested with the restriction enzyme Xmnl and probed with pERT87-15. The affected male is deleted (no signal). His sister who was pregnant has a deleted X and an X chromosome with the 1.6/1.2 allele. Her husband’s X chromosome contains the 2.8 allele. The fetus contains the husband’s X and the deleted X. B: Diagram of the four possible outcomes of the prenatal diagnosis. The fetus is a carrier female. (Courtesy of A. D. Roses.) |
orthoses or knee-ankle-foot orthoses combined with standing devices or walkers. Surgical release of contractures is only occasionally used for functional reasons, such as maintaining standing or ambulation. During the wheelchair years, surgical scoliosis stabilization can help maintain a comfortable sitting position and may reduce the risk of atelectasis and pneumonia.
The characteristic humeroperoneal distribution of weakness is unusual. That is, the biceps and triceps are affected rather than shoulder girdle muscles, and distal muscles are affected more than proximal muscles in the legs. The limb weakness may be mild or severe.
Contractures are disproportionately severe and are evident before much weakness is noted. The contractures affect the elbows, knees, ankles, fingers, and spine. A rigid spine develops, and neck flexion is limited.
Heart block, atrial paralysis, and atrial fibrillation are found in 95% of patients by age 30 years and lead to placement of a pacemaker. Dilated cardiomyopathy occurs in about 35% of all cases.
The name designates the characteristic distribution of weakness, which causes symptoms that differ from those of DMD. The face is almost always affected. Severity varies, and some of those affected are asymptomatic but show diagnostic signs. Progression is slow. Symptoms usually begin in adolescence, but signs may be evident in children. Serum CK levels are normal or minimally elevated.
Facial weakness is evident not only by limitation of lip movements but also in the slightly everted lips and wide palpebral fissures. Patients may state that they have never been able to whistle or blow up a balloon. Some are said by relatives to sleep with eyes open.
Scapular winging is prominent. Protrusion of the scapulae is more evident when the patient tries to push against a wall with elbows extended and hands at shoulder level. The winging becomes more evident when the patient tries to raise the arms laterally. The patient cannot raise the arms to shoulder level even though there is no weakness of the deltoids on manual testing. This limitation is caused by inadequate fixation of the scapulae to the chest wall.
The shoulder girdle has a characteristic appearance. Viewed from the front, the clavicles seem to sag and the tips of the scapulae project above the supraclavicular fossa. This abnormality becomes more marked when the subject tries to raise the arms laterally to shoulder level. Smallness of the pectoral muscles affects the anterior axillary fold, which is ordinarily diagonal but assumes a vertical position. Abdominal muscle weakness may cause abdominal protrusion and exaggerated lumbar lordosis. The lower abdominal muscles are weaker than the upper causing a positive “Beevor” sign with the navel moving toward the head upon neck flexion in a supine position. The limb muscle weakness is often asymmetric.
Leg weakness may affect proximal muscles or, more often, the tibialis anterior and peroneals, resulting in footdrop.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


