CHAPTER 328 Regeneration and Repair
Historically, neurosurgical interventions for traumatic brain injury (TBI) have centered on strategies to reduce sequelae of the primary brain insult. Despite progress in understanding the pathophysiology of TBI, clinical neuroprotection trials targeting these secondary injury mechanisms have failed to show consistent improvement in the outcomes of head-injured patients (at least 21 multicenter clinical trials have been conducted since 1985).1,2 In addition to local neuronal destruction resulting from focal injury, mechanical forces secondarily induce a progressive cascade of related events that contribute to neuronal death, including ischemia, brain edema, diffuse axonal injury (DAI), excitotoxicity, radical-mediated damage, mitochondrial dysfunction, and dysregulation of calcium homeostasis.3,4 In response to these processes, however, the injured brain activates a limited program for plasticity and cellular regeneration. Demonstration of these latent mechanisms in animal and human studies has spawned great interest in developing techniques to facilitate repair of the neuronal circuitry damaged by TBI.
Restorative strategies represent a shift in therapeutic goals from focusing solely on salvaging acutely threatened tissue to introducing interventions that support spontaneous and directed functional recovery (Fig. 328-1). Much of the evidence for spontaneous recovery of the damaged cerebrum comes from the stroke literature, where studies in animal models have provided cellular and molecular information, whereas systems-level data are increasingly being obtained from neuroimaging or neurophysiology studies in patients. Clinically, several basic principles of recovery have been indentified: most spontaneous recovery occurs within 3 to 6 months, cognitive deficits are more likely than motor deficits to show further gain beyond this point, the rate of recovery is inversely proportional to the severity of the deficit, and recovery patterns vary between types of deficits in the same patient.5 These observations suggest multiple mechanisms of recovery whose effectiveness varies with the type and location of the physical injury, as well as with the type and severity of the clinical findings. These mechanisms can be generalized to three basic categories: plasticity of intact networks, repair of damaged circuitry, and replacement of lost neurons. This chapter describes the opportunities and obstacles inherent in potential therapeutic strategies for repair and regeneration after TBI, including manipulation of endogenous neurogenesis, cell transplantation, gene therapy, neurorehabilitation, and electrophysiologic manipulation.
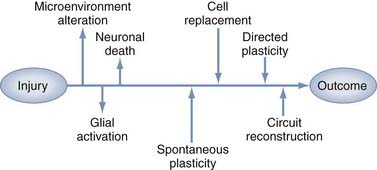
Trauma-Induced Plasticity
The brain activates a dramatic response to traumatic injury that is initially manifested as changes in the cellular microenvironment. These changes involve inflammation, disruption of physical structure and protein expression in the extracellular matrix (ECM), and altered expression of trophic factors. Although some of these mechanisms attenuate acute damage at the expense of future regenerative capacity, others retain the potential to participate in therapeutic interventions. Plasticity occurs when injury induces reorganization of cortical connections, but plasticity is not always synonymous with functional recovery. Even though processes such as synaptic sprouting, unmasking of dormant circuits, and the development of new polysynaptic connections can enable function, plasticity can also restrict function, as occurs when posttraumatic epileptic seizure foci or neuropathic pain is produced.6 This dichotomy is most evident in the developing brain, where increased plasticity has traditionally been thought to confer an advantage for recovery from TBI. Recovery also strongly depends on the type of injury,7 and the relationship between age and functional outcome is different within pediatric and adult age groups.8 These points reiterate the fact that plasticity is a complex series of molecular, cellular, and physiologic events that must be carefully orchestrated for both optimal maturation of the developing brain and optimal functional recovery from traumatic injury.9
The differences between injury responses in the developing and mature brain are also quite important when one considers that functional recovery after injury may require recapitulation of developmental events to produce neuronal circuit reconnections. Work in animal models has shown that focal damage in the adult brain can lead to a number of molecular and cellular changes, in both perilesional and remote brain regions, that are normally seen only in the developing brain.10 These distinctions should be kept in mind when considering how plasticity is altered by injury-induced microenvironmental changes in neurotransmission, gene and protein expression, cell death, neurogenesis, and neural connectivity.
The Injury Microenvironment: A Double-Edged Sword
In addition to local neuronal destruction resulting from the primary mechanical insult, TBI induces a progressive cascade of delayed secondary events that contribute to neuronal death, including ischemia, “wallerian degeneration” secondary to DAI, excitotoxicity, free radical–mediated damage, mitochondrial dysfunction, and dysregulation of calcium homeostasis. Injury may occur in the form of focal damage, as typically occurs after acute subdural hematoma or contusion, or it may be diffuse with widespread delayed neuronal loss, as typically occurs after DAI. Focal damage is characteristically seen around hemorrhagic lesions such as contusions within the gray matter or at gray-white matter junctions. These lesions are usually located at the frontal and temporal poles and in the orbital frontal cortex.11 Such focal neuronal death may occur as a result of both rapid necrotic and slower apoptotic mechanisms.12,13 Among diffuse injury sites, the hippocampus is known to be especially vulnerable in humans, with neuronal loss occurring in more than 80% of fatal TBIs, even in the absence of elevated intracranial pressure.14,15 After the acute phase of focal TBI and brain ischemic events, hippocampal neurons may be the most vulnerable neurons in the brain in that they show the earliest evidence of TBI-induced degeneration in experimental models.16 These hippocampal changes correlate with the profound memory impairment that is seen in both human and animal models of TBI.17,18 In the two most frequently used experimental TBI models, the lateral fluid percussion (LFP) and controlled cortical impact (CCI) injury models, selective neuronal death has also been well described in the hippocampus.19
The slow diffuse neuronal loss that is often seen in DAI, involving apoptotic morphology and wallerian degeneration, was previously thought to be an inevitable result of axonal interruption, as seen with primary axotomy of central nervous system (CNS) neurons. More recently, animal studies of DAI have shown that despite the proximity of traumatic axonal injury to the neuronal somata, neuronal cell bodies do not always show a pathologic progression to cell death and in fact some neurons and axons exhibit changes suggestive of reorganization and potential repair.20 These data suggest that injured neurons may be amenable to rescue through augmentation of endogenous plasticity. Therapeutic interventions, however, must account for changes in the injury microenvironment that encompass barriers to both functional reconnection and opportunities for repair (Table 328-1), as discussed in the following sections.
TABLE 328-1 Microenvironmental Factors and Potential Role in Neuroplasticity
| RESPONSE | DISABLING ROLE | ENABLING ROLE |
|---|---|---|
| Inflammation | Cytokine release Complement activation | Glial activation Microglial activation |
| Reactive gliosis | Neurite inhibition Dysmyelination | Neurotrophin secretion Neurotransmitter regulation |
| ECM deposition | Neuropil distortion | Growth permission |
| MMP activation | Synapse dysregulation | Substrate guidance |
| Neurogenesis | Seizure potential | Cell replacement |
ECM, extracellular matrix; MMP, matrix metalloproteinase.
Inflammatory Mechanisms and Gliosis
TBI produces a neuroinflammatory condition of the CNS in which rupture of the blood-brain barrier leads to accumulation of leukocytes from the systemic circulation and subsequent initiation of the immune functions of native glia.21 Acute inflammatory cytokine-mediated events such as monocyte/macrophage-mediated phagocytosis22 and complement-mediated cytolysis23 are now well recognized early after TBI and persist at least for several days and weeks. This response is partially represented in the form of “microglial stars” and “perivascular cuffing” in human pathologic specimens.24 Additionally, in a primate cortical lesion model, microglia and macrophages remain reactive in the cortex near the lesion and along sites of wallerian degeneration (corpus callosum and corticospinal tracts) for at least 12 months.25 The expression of brain-derived neurotrophic factor (BDNF) by a subpopulation of microglia/macrophages at this late time point suggests a possible neurotrophic role for microglia in long-term recovery.
Both inflammatory mechanisms and necrotic cell death trigger reactive gliosis, the astrocytic response to injury that consists of astrocyte hypertrophy, activation of fibrillary processes, and penumbral proliferation.26 Staining of glial fibrillary acidic protein (GFAP) increases dramatically in many areas of the brain days, weeks, and months after severe TBI.27 Upregulation of intermediate filaments, a hallmark of reactive astrocytes, was traditionally thought to be an obstacle to brain plasticity. In mice lacking GFAP and vimentin, however, the cellular processes of reactive astrocytes fail to exhibit the characteristic hypertrophy after entorhinal cortex lesions, as well as the initial increased loss of hippocampal neuronal synapses.28 Although these findings suggested that reactive astrocytes may play a beneficial role in the acute stage after a CNS insult, within a few days of injury, normal animals showed decreased synaptogenesis in comparison to animals lacking the two intermediate filament proteins, thus indicating a prohibitive effect of astrocytic hypertrophy at subacute stages.
The findings just presented support the idea that the beneficial effects of astrocytes at the site of an injury probably occur early in the injury response whereas subacute formation of the glial scar hinders regeneration. The beneficial effects of an astrocytic response include secretion of neurotrophic factors, regulation of metabolic factors (particularly important in times of stress), and maintenance of homeostatic levels of neurotransmitters.29 Astrocytes play a central role in neurometabolic coupling involving glutamate-stimulated aerobic glycolysis, a process that undergoes adaptations in parallel to synaptic plasticity.30 In both immature and adult TBI models, abnormalities have been reported in glutamatergic, GABAergic (transmitting γ-aminobutyric acid [GABA]), cholinergic, and aminergic transmission, whereas dysfunctional neurotransmission has been clinically implicated in pediatric memory impairment and attention problems.9 The detrimental effects of glial scars include inhibition of neurite outgrowth, inhibition of the remyelination of axons in white matter lesions, and hindrance of remyelination by inhibiting the differentiation of glial precursors into oligodendrocytes.29
Neural Connectivity
CNS neurons are intrinsically capable of axonal regeneration; however, this capacity rapidly diminishes postnatally at the onset of axonal myelination by mature oligodendrocytes. This loss of regenerative capacity can at least in part be attributed to the restriction of axonal regeneration and neuroplasticity by several CNS myelin proteins that inhibit neurite outgrowth, including Nogo-A, myelin-associated glycoprotein (MAG), and oligodendrocyte myelin.31 In neurons, Nogo-A is expressed during development and adulthood, whereas in oligodendrocytes, expression is initiated postnatally just before the appearance of myelin basic protein and myelination.32 With regard to TBI models, Nogo-A is upregulated after fluid percussion injury (FPI),33 and intraventricular infusion of an anti-Nogo antibody has been associated with improved postinjury cognitive recovery.34,35 Recently, it has become clear that the production of other inhibitory molecules, such as proteoglycans, is equally as important in creating a nonpermissive environment for axons attempting to regenerate.36
Mechanical obstruction of axonal regeneration by concomitant deposition of ECM molecules, which distorts the architecture of the neuropil, has also been well described. Conversely, levels of two ECM molecules critical to neural development, fibronectin and laminin, increase after human and experimental brain injury and may play a protective or reparative role in the injury response.37 The family of matrix metalloproteinases (MMPs) that regulate ECM molecules also performs essential functions during neuroplasticity in both developing and adult nervous systems, including substrate guidance during neuritogenesis and the establishment of boundaries for axonal terminal fields. In animals injured by entorhinal cortex lesions, MMPs regulate reactive synaptogenesis in a time- and injury-dependent manner to the extent that their inhibition interferes with emergence of the capacity for long-term potentiation in the sprouting crossed temporodentate pathway38 and produces deficits in spatial learning.39
The pervasive memory, cognitive, and motor deficits apparent in patients with mild to moderate TBI, sometimes occurring in the absence of frank neuronal cell loss, suggests that TBI alters the balance between excitatory and inhibitory neurotransmission in surviving neurons and disrupts the synaptic circuitry in the hippocampus.40 This effect occurs in a temporal and regional fashion such that in some pathways, injury may induce either increased excitability41 or inhibition42 of dentate gyrus granule cell neurons, thereby potentially affecting the ability of these cells to resist the synchronized bursting characteristic of seizure activity. In addition, the hippocampus ipsilateral to the cortical contusion is capable of a significant plasticity response, but synapse replacement in this area does not necessarily result in significant improvement in spatial learning.43
Gene and Protein Expression
Gene and protein profiling strategies have been used to study plasticity-related gene expression after experimental TBI. Several studies have shown that expression of neurotrophic factors is significantly altered after experimental TBI. RNA studies have demonstrated that some neurotrophic compounds such as nerve growth factor (NGF) and BDNF are upregulated44,45 whereas others such as NT-3 are downregulated.46,47 Data from the CCI model suggest that fibroblast growth factor 2 (FGF-2) is also upregulated, which stimulates posttraumatic neurogenesis and preserves granule cell layer volume, effects that are increased with FGF-2 supplementation via gene transfer.48 S100B is a neurotrophic and neuroprotective calcium-binding protein produced primarily by astrocytes that despite a correlation between increased cerebrospinal fluid levels and poor prognosis in TBI patients, has a beneficial effect on neuronal maintenance, neurogenesis, and cognitive performance in rodent TBI models.49
These alterations are not simple issues of upregulation versus downregulation because they occur with temporal, subregional, and age-related specificity. For instance, BDNF levels after experimental TBI are increased to a greater extent in aged animals than in young animals.50 BDNF is an activity-related molecule closely associated with experience-dependent plasticity and developmental regulation, and it may play a compensatory role after mild TBI in the developing rodent brain, where despite reductions in BDNF mRNA and protein expression in the ipsilateral cortex and hippocampus, contralateral upregulation occurs up to 2 weeks after injury.51 NGF-induced protein B, a transcription factor, is upregulated in a severity-dependent fashion in the immature brain, but it is increased independent of severity in adults. In contrast, the synaptic activity–inhibiting, calcium-binding protein synaptotagmin IV increases only in developing animals after severe injury and not at all in adults.52 Although these examples illustrate the complexity of the molecular injury response, they also suggest many opportunities to improve our understanding of important differences between developmental and injury-related plasticity.
Regional intrinsic differences in gene expression before and after injury are also likely to define the pathology and recovery of function after TBI. The differential vulnerability of the hippocampus to TBI as compared with the high degree of plasticity of the cortex, which may underlie the reason why memory dysfunction lingers in comparison to motor and sensory recovery, has been suggested by the fact that the majority of mRNA molecules that change after injury were found to be exclusively altered in either the hippocampus or the frontal cortex.53 Within the hippocampus itself, gene expression appears to be influenced by both injury severity and time since the insult. Many genes encoding molecules for cellular signaling, synaptic plasticity, metabolism, ion channels, and transporters that are initially upregulated after severe injury are downregulated after moderate injury, although moderate injury is associated with an increasing number of responsive genes as a function of time after injury as opposed to a decreasing number after severe injury.54 Overall, there is still much to discover about region- and time-specific microenvironmental dynamics, all of which combine to form various posttraumatic brain niches.
Endogenous Neurogenesis after Traumatic Brain Injury
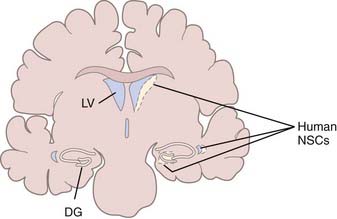
TBI induces changes in the specialized microenvironment that regulates the proliferation of neural stem cells (NSCs) and differentiation of neuronal progenitor cells (NPCs). These neurogenic niches are found in the human dentate gyrus of the hippocampus and the subventricular zone (SVZ) of the lateral ventricular walls (Fig. 328-2).55 The best-characterized neurogenic region in the adult human brain is the SVZ lining the lateral ventricle, where a ribbon of SVZ astrocytes have been identified that proliferate in vivo and behave as multipotent progenitor cells in vitro.56 Unlike the situation in rodents and primates, where neurons born in the SVZ migrate in chains through the rostral migratory stream to replace interneurons of the olfactory bulb,57–59 there is no evidence of chains of migrating neuroblasts in the human SVZ.60,61 It has been estimated that in normal humans, less than 1% of astrocytes within the SVZ ribbon are in division,56 and although these endogenous NSCs can be expanded in culture,62 their response to injury in patients has not been studied. Less well characterized in the human brain are proliferating NPCs in the hippocampus,63 which have also been demonstrated to be multipotent progenitor cells in vitro.64
Rat models have been used to investigate TBI-induced neurogenesis and have shown that the number of NPCs in both the SVZ and hippocampus are substantially increased after TBI (Fig. 328-3), a process that is more robust in juvenile animals.65 Newborn NPCs from the SVZ appear to have the potential to migrate to areas of focal cortical damage, whereas hippocampal neurogenesis occurs in the setting of diffuse local damage. Nevertheless, in adult and aged animals, the number of new neurons developing after TBI remains small in comparison to astrocyte and oligodendrocyte differentiation, as assessed by bromodeoxyuridine (BrdU) staining.
Subventricular Zone Neurogenesis after Trauma
In common with models of seizure, ischemia, and deafferentation injury, experimental TBI induces an increase in SVZ NPC proliferation and a secondary redirected streaming of these precursors into the injured zone.66 Studies demonstrate up to twofold increases in ipsilateral SVZ NPC proliferation within the first 2 to 5 days after FPI67–70 and as much as a fourfold increase after the more focal CCI up to 14 days after injury.71,72 Contralateral NPC proliferation in the SVZ also increases, apparently to a lesser extent, after injury in both models.69,70,72 Newborn cells colabeled with BrdU and neuronal phenotypic markers have been observed in the cortex, striatum, and corpus callosum as long as 45 days after injury. Patch clamp studies have demonstrated that such BrdU cells exhibit neuronal electrophysiologic properties,73 and anatomic integration of these new neurons into host tissue has been shown by retrograde tracer labeling and synaptophysin triple-label immunohistochemical techniques.74
Migration of Subventricular Zone Neural Progenitor Cells to the Site of Injury
Glial cell proliferation has long been appreciated at sites of focal TBI damage, but more recently, it has been shown that some glia migrate into the lesion site from the SVZ and that these cells arise from NPCs. After the intraventricular injection of fluorescent microspheres to label BrdU-positive subventricular cells, migration from the SVZ to the cortex surrounding a cortical contusion impact site has been observed.71 In the SVZ, corpus callosum, striatum, and cortex, many of these cells expressed doublecortin, a marker of immature neuroblasts, thus suggesting that local neurogenesis could be driven by injury-specific extracellular signaling pathways. In the pericontusional cortex, expression of the mature neuronal marker NeuN was seen in cells tagged before injury by intraventricular infusion of a lipophilic dye, a finding suggesting that these cells were derived from SVZ NPCs that migrated to the site of injury.75
Hippocampal Neurogenesis after Brain Trauma
Although selectively more vulnerable to TBI, the injured hippocampus may have a unique ability to locally replace damaged neurons inasmuch as the dentate gyrus is normally an area of active endogenous neurogenesis. In humans, in vivo dentate gyrus neurogenesis was demonstrated on histologic sections obtained from patients who had been administered BrdU for diagnostic purposes.63 Studies from rodents also demonstrated that cells with an astrocyte phenotype residing in the adult subgranular zone of the dentate gyrus continually generate cells with a neuroblast phenotype that migrate into the granule cell layer.76 Although some of these new neurons die by apoptosis, many of these new neurons demonstrate functional integration within the hippocampal circuitry by sending dendrites into the molecular layer and axons into the CA3 region,77,78 and they additionally exhibit mature neuronal electrophysiologic characteristics.79
Moderate and severe TBI is always associated with learning and memory deficits—the “functional hallmarks” of TBI. This is usually accompanied by histologic damage in the form of cell loss, especially in the dentate gyrus and CA1 layers. Because neurogenesis in this region has been linked to enhanced learning and memory functions,80 it is reasonable to hypothesize that injury-induced neurogenesis may function in the replacement of damaged neurons and thus contribute to repair of neuronal circuits and restoration of neurological function in patients with TBI. After LFP, cell proliferation increases in the rodent dentate gyrus threefold to fourfold beginning as early as 2 days after injury, peaks during the first week after injury, and returns to baseline levels by day 35 after injury.67,81 Similarly, CCI may induce up to a sixfold increase in dividing cells in the ipsilateral dentate gyrus and a significant but smaller increase in the contralateral dentate gyrus.48,72,82–84 A significant increase in the number of new cells that express neuronal markers has been reported after longer maturation periods. TBI can initially induce a fourfold increase in the number of BrdU-labeled cells that colabel for NeuN, a mature neuronal phenotypic marker, nearly half of which migrate to the inner half of the granular cell layer and persist 10 weeks after injury.74 Others have reported a fivefold increase in the number of injury-generated double-labeled neurons, seen as late as 60 days after injury.84 The central issue remains, however: do these new neurons functionally integrate with the host hippocampal tissue and exhibit appropriate electrophysiology?
To begin to answer these questions, hippocampal neurons born after injury, which express NeuN and calbindin, have been labeled by stereotactic retrograde tracer injections of the neuronal tracer fluorogold into the CA3 region, which is one target zone for the granular cell neuronal projections.74 After 2 weeks, about 30% of the BrdU-positive cells in the dentate gyrus were labeled with fluorogold, a finding suggestive of connectivity with the CA3 target region, and these double-labeled cells were increased fourfold by FPI. Labeled cell bodies were also frequently enveloped by a synaptophysin “lattice,” as described previously,78 thus suggesting the formation of synapses onto the somata of newborn neurons.
In terms of measuring whether posttraumatic neurogenesis is functional on a broader scale that affects behavior, memory function, as assessed by Morris water maze tasks, is decreased in association with procedures that abolish the proliferation of new cells, such as the administration of antimitotic drugs.84a Low-dose irradiation, which has been shown to markedly reduce neurogenesis, also significantly reduces the ability to recover spatial memory function after experimental TBI.85 Additionally, inactivation of a subset of adult hippocampal NSCs in mice was shown to produce both a significant reduction in overall stem cell proliferation and a selective decrement in spatial learning.85a
Cortical Neurogenesis after Brain Trauma
Although the hippocampus is selectively damaged in TBI, the greatest density of neuronal loss occurs with focal parenchymal contusions arising in locations that vary with the heterogeneity of both the primary injury and subsequent mechanism of secondary injury. As discussed earlier, the injured brain may induce migration of neuroblasts from the SVZ into cortical lesions, but latent NPCs may potentially be activated at sites of cortical injury in the mammalian brain. In songbirds such as zebra finches, it has long been known that some cortical projection neurons are lost and regenerated each year from SVZ precursors in response to evolutionary pressure.86 Macklis and colleagues suggested the possibility that endogenous neural precursors from the cortex itself in rodents may differentiate into corticothalamic neurons in layer VI of the anterior cortex after targeted neuronal death and survive at least long enough to form appropriate long-distance corticothalamic connections.87 In humans, the existence of such NPCs in nongerminal cortex has been suggested from some isolation and culture studies, including the isolation and continuous culture of putative neuroblasts from neocortical tissue surgically resected from TBI patients at the time of decompressive craniotomy for high intracranial pressure,88 as well as the isolation of neurosphere-generating cells from similar cortical tissue.89 After CCI in rodents, the formation of neurospheres from neocortical cells has also been observed, but only when the injured tissue was isolated at 3 days after injury, which corresponds to a peak in nestin expression in pericontusional cortex.90 Induction of nestin expression in the cortex adjacent to the CCI injury has also been reported at 7 days after injury,84 thus suggesting the possible activation of resident cortical NPCs, although in both cases these cells could alternatively have migrated from the SVZ.
Effect of Age on Posttraumatic Neurogenesis
Juvenile mammals usually recover cerebral function to a greater extent than adults do after TBI. Because cell proliferation in the dentate gyrus decreases with age in normal rats,91 it has been hypothesized that age-related differences in hippocampal neurogenesis after TBI may be responsible in part for the greater cognitive recovery typically seen in young mammals.81 Juvenile rats generate nearly twice the number of new cells in the subgranular zone than adults do in the first 2 days after LFP injury, although this response appears to be related to a greater level of basal juvenile neurogenesis because the percent increases in cell number above baseline are similar in young and old animals. Additionally, more newly generated cells appear to differentiate into mature neurons over time in juveniles. Typical granule cell neurons have been shown to functionally integrate by 14 days after generation in the normal adult rodent brain77 and become critically involved in the learning response within this same period.92 Fewer cognitive deficits and better recovery are observed during the first 2 weeks after injury in juvenile rats than in adults.93 This difference may therefore result in part from a greater ability of younger animals to generate new hippocampal granule cell neurons in response to injury. The mechanism by which neuronal differentiation is enhanced in younger animals is not clear, but answers to this question may aid discovery of therapeutic interventions to optimize neurogenesis after TBI.
Despite a constant decline in NPC proliferation in the dentate gyrus from adolescence through senescence,91,94,95 the adult brain upregulates proliferation to almost the same extent as the juvenile brain after TBI, thus indicating that NPCs conserve the ability to respond to proliferative signals. Although injury signals increase the proliferative rate of NPCs, the increased presence of actual newly generated neurons in younger brains may reflect greater integrity of the neurogenic niche mechanisms required for differentiation or survival of new neurons. One possible mechanism, similar to that hypothesized for the effects of an enriched environment on hippocampal neurogenesis (a survival-promoting effect of newborn cells that is selective for neurons),96 is a greater level of specific neurotrophin support97 that is either not present or not induced in adults. All of the putative mechanisms just described, however, would be nearly irrelevant without clinical evidence of functional recovery in the human brain after TBI.
Evidence of Functional Recovery
Spontaneous recovery from TBI varies with the heterogeneity of the injury, and consequently, a wide range of outcomes are produced: in some vegetative patients, functional magnetic resonance imaging (fMRI) demonstrates appropriate cortical responses to spoken commands that are indistinguishable from normal responses,98 whereas in patients with mild TBI, diffusion tensor imaging demonstrates variations in microstructural white matter integrity that are associated with persistent impairment in memory and attentional control.99 Approximately 85% of recovery from TBI occurs within 6 months after injury,100 and functional neuroimaging techniques have increasingly been used as a starting point to understanding the appropriate anatomic correlates.
Functional Imaging
The molecular events described in the previous section are difficult to measure in TBI patients, and therefore functional neuroimaging techniques have been used to investigate tissue function and neurophysiologic responsiveness after injury. Most TBI-related studies to date have involved fMRI of stroke patients. These studies suggest that the behavioral improvements seen in the weeks after injury are supported by compensatory reorganization of surviving neurostructural elements as follows: increased activity in brain regions distant from but connected to the injured zone, increased activity in the contralesional hemisphere (reduced laterality), and shifts in representational maps within intact cortical regions surrounding the injury zone.5 Each of these events may be a target for therapeutic intervention, although stroke studies assessing sensorimotor, language, and right hemisphere attentional deficits suggest that the final behavioral outcome is related to the degree of activity in the corresponding primary cortex.5
The motor system has been studied more extensively than other systems as a model for basic plasticity phenomena because control of motor behavior is so easily evident, but understanding the neurophysiology of motor function and the integral role of sensory input in motor control is fundamental for understanding general plasticity mechanisms.101 Transcranial magnetic brain stimulation (TMS) is used to study brain plasticity by applying a brief and intense magnetic field directly to the scalp and recording motor evoked potential responses to somatotopically map the cortical motor output. Magnetoencephalography is an additional imaging technique that spatially identifies the synchronous firing of neurons from restricted cortical areas in relation to either spontaneous cerebral activity or external stimuli, thereby allowing precise three-dimensional localization of the firing neuronal pool. By using these methods, it is possible to demonstrate lesion- or use-dependent changes in cortical maps. Combined-modality functional imaging studies suggest that reorganization of motor output is ongoing for several months after brain injury, and the mode and degree of motor recovery may largely depend on functional compensation by spared parallel descending pathways, activation of the contralateral premotor cortex, and cerebellar activation.101






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


