Rufinamide
Tracy A. Glauser
Meir Bialer
Introduction
Rufinamide is a broad-spectrum antiepileptic drug (AED) with documented ability to block sodium (Na) channels.24,34 Following discovery of antiepileptic activity in animal models, it has been undergoing Phase II and III clinical trials. Rufinamide has demonstrated efficacy against partial seizures in adults and generalized seizures in patients with the Lennox-Gastaut syndrome (LGS).5,10,14 Clinical trials have reported a good tolerability profile. Rufinamide is currently under evaluation by the European Medicines Agency and the United States Food and Drug Administration (FDA).
Chemistry
Rufinamide (1–(2,6–difluorophenyl)methyl-IH-I,2,3triazole-4–carboxamide) (Fig. 1) is a triazole derivative structurally dissimilar to currently marketed AEDs.2,17 Rufinamide is a lipophilic compound with a partition coefficient (logP = 0.88) and solubility in water and in gastric and intestinal fluids of 40 to 70 mg/L.9 There are no currently marketed formulations of this compound. In clinical trials, film-coated tablets have been used.9
Plasma concentrations for rufinamide and its inactive carboxylic acid major metabolite (Fig. 1) are determined using high-performance liquid chromatography (HPLC).27 The limit of quantification of this validated HPLC assay is 25 ng/mL for rufinamide in plasma, 2.5 μg/mL for rufinamide in urine, and 5 μg/mL for rufinamide’s carboxylic acid major metabolite in urine.27
Pharmacology
Activity in Experimental Models of Seizures/Epilepsy
Rufinamide exhibits broad-spectrum anticonvulsant properties at nontoxic doses.34 In animal models, rufinamide’s protective index (PI = TD50/ED50) was superior to AEDs such as phenobarbital, phenytoin, ethosuximide, and valproic acid.2,34
The maximal electroshock (MES) test can identify AEDs with potential efficacy against partial seizures and generalized tonic–clonic seizures.20,22,26 Rufinamide given orally was protective against MES-induced tonic–clonic seizures in both mice and rats with an ED50 of 23.9 mg/kg and 6.1 mg/kg, respectively. Rufinamide’s anticonvulsant potency in the MES test was comparable to standard AEDs (Table 1).9 In rats and mice, tolerance was not noted over 5 days.34
The subcutaneous pentylenetetrazole (PTZ) test can identify AEDs with potential efficacy against clonic or absence seizures.20,22,26 Rufinamide’s efficacy in the subcutaneous PTZ test was species and route dependent. In mice, rufinamide suppressed PTZ-induced seizures with an ED50 of 45.8 mg/kg.34 Although orally administered rufinamide’s ED50 is less potent than phenobarbital’s ED50 (12.6 mg/kg), it is lower than the ED50 values for ethosuximide, valproic acid, and phenytoin9 (Table 2). Intraperitoneal rufinamide suppressed PTZ-induced clonus with an ED50 of 54.5mg/kg in mice.34 In rats, oral rufinamide was inactive against subcutaneous-induced PTZ seizure9 (Table 2).
Intraperitoneal rufinamide blocked clonic seizures induced by subcutaneous bicuculline and picrotoxin in mice with ED50 values of 50.5 mg/kg and 76.3 mg/kg, respectively.34 Similar to its results in the PTZ test, rufinamide was less potent than phenobarbital but more potent than ethosuximide and valproic acid (phenytoin is inactive in this test).9 For seizures induced by intraperitoneal picrotoxin, rufinamide had lower potency (ED50 of about 300 mg/kg). Rufinamide’s lowest potency was seen in mice in the glycine-related subcutaneous or intraperitoneal strychnine test, where the maximum protection rate of 37.5% occurred at a dose of 125 mg/kg.9,29 Overall, rufinamide’s tolerability (determined by either the chimney test in rats or the Rotorod test [6 rpm] at time of peak neurotoxic effect in mice) was similar to or exceeded that of any established AED except phenytoin administered orally to rats.34
Once-daily threshold electrical stimulation of the amygdala in animals induces epileptic electroencephalogram (EEG) afterdischarges and results in the kindling phenomenon.15,16,20,28 In rats treated daily with oral rufinamide (20 mg/kg and 60 mg/kg) an increase in afterdischarge duration occurred, but no change in convulsive behavior was seen compared with controls.29 Carbamazepine and phenytoin produce similar effects.23,30 In cats, oral rufinamide (100 mg/kg and 300 mg/kg) delayed kindling development and suppressed afterdischarges in fully kindled animals. Carbamazepine (40 mg/kg) and valproic acid (180 mg/kg) produced similar effects.9 Rufinamide antagonized kindling but did not provoke motor disturbances.9
Electrically induced hippocampal and cortical afterdischarges are also used to investigate the effects of AEDs against seizures originating from a focus. In cats, intraperitoneal rufinamide (300 mg/kg) reduced hippocampal and cortical afterdischarge duration by more than 50%.9 Rhesus monkeys with aluminum hydroxide implants in the motor cortex provide an animal model of chronically recurring partial seizures with or without secondary generalization. Subchronic treatment with oral rufinamide (30–50 mg/kg daily for 15 days) reduced seizure frequency by 75% to 100% without a significant change in mean seizure duration.9
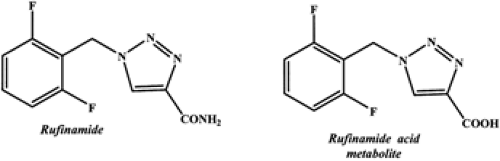 FIGURE 1. Chemical structure of rufinamide and its major acid metabolite. |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
Mechanism of Action
Rufinamide’s full mechanism of action is not yet known in detail. Some of its anticonvulsant effect is thought to result, in part, from blocking Na channels.24 In rat cortical neurons, over a wide range of concentrations, rufinamide caused rest- and use-dependent block of Na currents coupled with slowed recovery from inactivation.24 The rufinamide concentration that limited firing in 50% of neurons was 3.8 μmol /L (range 2–10 μmol/L).24
Rufinamide does not significantly interact with neurotransmitter systems, including γ-aminobutyric acid (GABA), adenosine, monoaminergic and cholinergic binding sites, N-methyl-D-aspartate (NMDA), and other excitatory amino acid binding sites.17,29
Rufinamide does not significantly interact with neurotransmitter systems, including γ-aminobutyric acid (GABA), adenosine, monoaminergic and cholinergic binding sites, N-methyl-D-aspartate (NMDA), and other excitatory amino acid binding sites.17,29
Clinical Pharmacokinetics
Absorption
During Phase I clinical trials, single- and multiple-dose studies were performed in healthy subjects to assess the pharmacokinetic profile of rufinamide. Based on urinary recovery of radioactivity following oral administration of C14–labelled drug (600 mg as capsules) with food, the bioavailability of low doses of rufinamide was estimated to be approximately 85%.9 Peak plasma concentration (Cmax) of rufinamide was reached 5 to 6 hours after dosing.2,3,7 The intersubject variability in the mean area under curve (AUC) and Cmax values of rufinamide were less than 26% for both tablets and suspension. Contribution of the intrasubject variability to the overall variability was small (20%).2,7 The tablet formulation was bioequivalent to the suspension in terms of rate and extent of absorption. Rufinamide exhibited dose-limited absorption; doses higher than 400 mg for both fasted and fed healthy subjects demonstrated less than proportional increase in peak concentrations (Cmax) and plasma AUC.9 Similar to the single-dose healthy volunteer studies, multiple-dose studies found a less than dose proportional increase in steady-state rufinamide plasma levels, due to dose-limited absorption.9
After intake in the fasting state, rufinamide has an oral bioavailability of approximately 60%; however, food increases rufinamide’s extent of absorption and plasma exposure (AUC).3,15 In a 1998 Phase I study using an older formulation, food increased the extent of absorption or plasma exposure AUC by 44% and the Cmax by 100%.6 The time to reach Cmax (tmax) was 8 hours in fasted conditions and 6 hours in those fed after breakfast.6 However, unpublished data using a newer formulation of rufinamide indicates food increases rufinamide’s extent of absorption or plasma exposure AUC by 34% and the Cmax by 56%.9 The increase in rufinamide’s AUC when administered concomitantly with food might be due to a change in gastrointestinal absorption and dissolution of the parent drug. The low water solubility and high lipophilicity (as indicated by its relatively large partition coefficient; logP = 0.88) supports the hypothesis of an increased solubility in the presence of food due to a larger volume of liquid and stimulated biliary secretion.5
Plasma Protein Binding and Distribution
Rufinamide’s protein binding is low (34%).2,17 Binding is predominantly to albumin. Rufinamide is evenly distributed between erythrocytes and plasma, and its apparent volume of distribution (V/F) is similar to the total body water (50–80 L).3,9
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


