Simple Partial Status Epilepticus and Epilepsia Partialis Continua of Kozhevnikov
Heinz Gregor Wieser
Patrick Chauvel
Introduction
Literature reports of simple partial status epilepticus (SPSE) seem to be relatively rare in comparison with those of generalized convulsive SE, complex partial SE (CPSE), and absence SE. One reason may be that SPSE very often progresses to CPSE, so that many overlaps between SPSE and CPSE exist in the literature, and cases are finally categorized according to their full-blown semiology, that is, as CPSE, even though for a certain time they might fulfill the criteria of SPSE (for definitions, see later discussion). Moreover, some subjective forms of SPSE have been described under the recently rediscovered term “aura continua”285 and nonconvulsive SE (NCSE),121 although levels of alteration of consciousness in the latter are often difficult to assess.
Among the multiple potential subtypes of SPSE, certain manifestations (which are readily accessible to the patient’s awareness or physician’s observation) seem to predominate, such as SPSE with motor symptoms and with certain sensory symptoms. Numerous studies have dealt with the pathophysiology of epilepsia partialis continua (EPC), whereas few have been devoted to SPSE with the expression of emotional/affective and subtle vegetative-autonomic symptoms only. Apart from the problem of the so-called interictal personality and behavioral syndrome271 and other described peculiarities of personality, there remains the question of the boundaries of SPSE being expressed through very localized, ongoing epileptic discharges in “relatively silent” brain regions (e.g., nondominant frontal lobe) that might not be associated with any overt symptom at all but might be detected only with subtle decreases in very sophisticated neuropsychological test performance.
In contrast to the history of epilepsy, SE was infrequently recorded until the dissertation of Louis Calmeil,40 where the expression état de mal is first found, but still in reference to convulsive generalized SE only. The proceedings of the Xth Marseilles Colloquium of 1962 represent the first monograph (Les états de mal épileptiques)94 on this subject. At the Marseilles Colloquia of 1962 and 1964, definitions and classifications of seizures and of SE were proposed with the obvious notion that there are as many types of status as there are types of seizures.
The modern conceptual basis of SE as having an “etymologic definition” and the notion that this term is to be used “whenever a seizure persists for a sufficient length of time [subsequently defined as at least 30 minutes] or is repeated frequently enough to produce a fixed or enduring epileptic condition” has been widely accepted and enshrined in the WHO Dictionary of Epilepsy90 as well as in the Handbook of Clinical Neurology210 and the Handbook of Electroencephalography and Clinical Neurophysiology.95 The first international classification of seizure types88,89 and its revision58 divided partial seizures into “simple” and “complex” according to whether consciousness was retained or lost. The subsequent classification of epilepsies and epileptic syndromes59 included a few syndromes that might conform to the widened definition of status (e.g., EPC, CSWS, Landau-Kleffner) but otherwise lacked a synoptic view. A major review of SE occurred at the Santa Monica, California, conference in 1980, which was published in 1983.66
In the new proposal,74 continuous seizure types are divided into generalized status epilepticus (including generalized tonic–clonic SE, clonic SE, absence SE, tonic SE, and myoclonic SE) and focal status epilepticus (epilepsia partialis continua of Kozhevnikov,284 aura continua,285 limbic SE [psychomotor status283], and hemiconvulsive status with hemiparesis). The division of partial seizures into “simple” and “complex” is not retained. In addition, the terms “partial” and “localization-related” have met with criticism, and a task force proposed that these terms be replaced by the older term “focal.”
Pathophysiologically, a clear distinction should be made between the terms epilepsia partialis continua (EPC), or so-called Kozhevnikov syndrome, and Rasmussen encephalitis (see section Pathophysiology). Clinically, Bancaud’s EPC type 2 largely corresponds with Rasmussen encephalitis.
Epilepsia partialis continua was first described by Kozhevnikov in 1894 as a “peculiar form of cortical epilepsy” that consisted of localized continuous jerks intermingled from time to time with spreading jacksonian seizures.129,130 In his 4 cases, the seizure disorder continued for 3.5 to 5 years in the same part of the body. Kozhevnikov postulated a localized inflammation of the brain involving the motor strip. Omorokow reviewed 42 cases of “Kozhevnikov syndrome” in the literature and described a further 52 cases from his Siberian clinic, recognizing that this form of epilepsy may be due to Russian spring-summer tick-borne encephalitis.174,175 In 1958, Rasmussen et al.206 described 3 cases of persisting focal epilepsy due to chronic focal encephalitis. By 1988, a total of 48 Rasmussen chronic encephalitis cases had been identified and described,205 differing in many ways from those of Russian spring-summer tick-borne encephalitis.7 Since the first descriptions of EPC, many localized cerebral disturbances have been found to give rise to similar patterns of ictal behavior,219 and the definition and nomenclature of EPC has become a subject of controversy.29 In 1966, Juul-Jensen and Denny-Brown120 pointed out that in various papers, the definition of EPC was not the same. They asked for a differentiation between true EPC with “clonic muscular twitching repeated at fairly regular short intervals in one part of the body for a period of days or weeks” and “focal epilepsy with motor seizures with frequent recurrence and with ‘Jackson march’ or progression from tonic to clonic phase.” Gastaut and Broughton92
commented that “certain authors now also include chronic localized myoclonus without intercurrent somatomotor seizures or, conversely, recurrent somatomotor seizures without local myoclonus. Other authors also include the subacute episodes of localized myoclonus observed during certain syndromes of diffuse hemispheric ischemia.” Later authors characterized EPC as a “partial somatomotor SE for a minimum of 1 hour and recurring in intervals of no more than 10 seconds”249 and as “spontaneous regular or irregular clonic twitching of cerebral cortical origin sometimes aggravated by action or sensory stimuli (reflex component) confined to one part of the body and continuing for a period of hours, days, or weeks.”171 In 1989 the International League Against Epilepsy (ILAE) Commission defined EPC (Kozhevnikov) syndrome as a specific form of partial somatomotor seizure disorders involving the rolandic area of the motor cortex.59 Schomer used EPC and focal SE as synonymous terms.219
commented that “certain authors now also include chronic localized myoclonus without intercurrent somatomotor seizures or, conversely, recurrent somatomotor seizures without local myoclonus. Other authors also include the subacute episodes of localized myoclonus observed during certain syndromes of diffuse hemispheric ischemia.” Later authors characterized EPC as a “partial somatomotor SE for a minimum of 1 hour and recurring in intervals of no more than 10 seconds”249 and as “spontaneous regular or irregular clonic twitching of cerebral cortical origin sometimes aggravated by action or sensory stimuli (reflex component) confined to one part of the body and continuing for a period of hours, days, or weeks.”171 In 1989 the International League Against Epilepsy (ILAE) Commission defined EPC (Kozhevnikov) syndrome as a specific form of partial somatomotor seizure disorders involving the rolandic area of the motor cortex.59 Schomer used EPC and focal SE as synonymous terms.219
Definition
For the purpose of this chapter, we suggest the following operational definition: SPSE is an epileptic condition (defined by clinical and electroencephalographic signs and symptoms) of at least 30 minutes’ duration with a large spectrum of clinical manifestations and encompassing subtle clinical signs, as well as at least some behavioral disturbances and psychosis-like states, in particular, elementary and complex (polymodal) hallucinations, without loss or severe alteration of consciousness (in other words, “with preserved consciousness” in the sense of an [operationally defined] adequate reactivity and response). Furthermore, we add the criterion that observed symptoms should fit with the known functional anatomy and, thus, with localization of the electroencephalographic (EEG) discharge, and, in the case of more diffuse and difficult-to-describe personality and behavioral changes, that there be a clear-cut relationship in time of the particular subtle signs and symptoms with epileptic EEG activity.
Epidemiology
Incidence
There exist no population-based studies on SPSE. Until very recently, computations of frequency were restricted to generalized tonic–clonic SE, but even here, only vague guesses could be given, at best. At a conservative and approximate estimate, status syndromes, including West syndrome, neonatal and childhood status syndromes, EPC, other SPSE, and absence status in the elderly, contribute to the annual incidence of status no more than 5 to 10 cases per one million persons in the general population. SPSE is, thus, very rare, but this statement might require revision if the boundary syndromes (mentioned earlier) are included.
Prevalence
Hauser112 attempted to break down SE by seizure type into the categories generalized (23%–51%), unilateral (39%), partial motor (24%–72%), partial, secondary generalized (5%–62%), other (5%), and nonconvulsive (5%–33%), based on the studies of Aminoff and Simon,5 Celesia,42 Hauser,111 Aicardi and Chevrie,2 and Forster et al.81 Unfortunately, no precise mention is made in these studies of the incidence and prevalence (or definite absence) of SPSE. Brett35 reported 22 children with “minor epileptic status” and differentiated this from “petit mal status” by the presence of myoclonus and less frequently occurring spike-wave patterns in EEG.
There seems to be a consensus that SPSE with motor phenomena is at least as common as SPSE without motor phenomena, that is, SPSE with sensory and/or autonomic signs and symptoms and SPSE with disturbances of language and higher cortical functions.
Epilepsia partialis continua is a condition with a wide range of underlying pathologies. Articles about EPC include case reports or small series of patients. For this reason, no epidemiologic data exist. Based on their survey of all registered cases in the United Kingdom during 1993, Cockerell et al.53 estimated the prevalence of EPC to be <1 per million.
Etiology
No specific etiology leading to SPSE is known, and a large spectrum of underlying causes has to be considered (See also section Differential Diagnosis).
Myoclonic seizures progressing into status (“myoclonic storm”), in general, may be due to a wide variety of etiologies (essential hereditary myoclonus epilepsy, lipidosis of the central nervous system, acute encephalitis, subacute sclerosing panencephalitis, acute cerebral anoxia, Creutzfeldt-Jakob disease, and renal insufficiency with uremia) and may occur in various epileptic conditions (common generalized epilepsy, Lennox-Gastaut syndrome, infantile spasms).167 Benign idiopathic neonatal convulsions, described in the literature as “fifth day fits,” present often as lateralized clonic seizures and may lead to a SE lasting about 20 hours (from 2 hours to 3 days).199 Some subtypes of the early infantile epileptic syndromes with suppression-burst must be considered here also. They can be viewed as falling into two main categories: (a) the Aicardi syndrome,1,3 which is, in essence, a complex malformation syndrome, and (b) an inborn metabolic disease, nonketotic hyperglycinemia.63 The majority of these patients have an EEG showing suppression-burst, for example, 98 of 146 in the Aicardi syndrome.50 Suppression-burst was also described in other metabolic or malformation syndromes, such as olivary dysplasia,109,207 hemimegalencephaly,177,250,267 Menkes disease,56,194 D-glyceric acidemia,100 methylmalonic acidaemia,152 propionic acidaemia,268 and sulfite oxidase deficiency.17 In early myoclonic encephalopathy (neonatal myoclonic encephalopathy), segmental and erratic myoclonias are usually the earliest ictal events (accompanied later by one or more of the other seizure types, such as massive myoclonias, simple partial seizures, and infantile spasms of the tonic type) and can recur frequently, so that they present as a status.1
Cases of epilepsia partialis continua (EPC) or Kozhevnikov syndrome have been a posteriori attributed to Russian spring-summer encephalitis.219 A Russian spring-summer tick-borne virus has been isolated from monkey brain after several months of incubation, supporting the hypothesis of a slow-viral disease.304 Dereux,67 reviewing a series of 102 cases of Kozhevnikov syndrome, found that >50% were caused by an “encephalitic process,” as were 31% of 85 pathologically verified cases in the review published by Löhler and Peters in 1974.141 In a study of 36 cases by Cockerell et al.,53 7 patients had Rasmussen encephalitis, 9 had vascular diseases, 4 had a “multisystem” disease, 4 had neoplastic diseases, 2 had perinatal injury, 1 had trauma, 1 had hyperglycemia, and 1 had Creutzfeldt-Jakob disease.
Rasmussen encephalitis roughly corresponds to Bancaud’s type 2 epilepsia partialis continua.19,20,21 In the statistics published by Oguni et al.,172 EPC developed in only 56% of the cases of Rasmussen syndrome as they evolved. In a recent Indian study of 20 patients,180 type 1 and type 2 EPC were found in respectively 55% and 45%; infectious etiologies were found in 60% (Rasmussen encephalitis in 25% of these, or 3 cases) and vascular etiologies in 25%, with the remaining 15% consisting of 1 mitochondrial disorder and 2 undetermined causes.
Creutzfeldt-Jakob disease (CJD) rarely presents with epilepsia partialis continua with subacute progression in which the EEG shows focal epileptiform activity and neuroimaging is negative.183
Patients with human immunodeficiency virus (HIV) infection may present new-onset EPC as an early manifestation of progressive multifocal leukoencephalopathy (PML).25,79
Cerebral tumors, such as astrocytomas, hemangiomas, lymphomas, and metastases, have been reported as causes of EPC. In fact, these tumors are associated with EPC because of their anatomic location near the central sulcus and probably not because of their neoplastic nature. The same relation may exist in cases of EPC in multiple sclerosis115,240,244 and in the earlier American120 and recent Indian cases180 with infectious mass lesions, especially of tuberculosis.
The vascular etiologies reported were occlusive, embolic, or hemorrhagic stroke; arteriovenous malformations; cortical venous thrombosis; and vasculitis in lupus and in Sjögren syndrome.23
Cortical dysplasias, especially hemimegalencephaly, can present with EPC, as well as tuberous sclerosis, linear sebaceous nevus syndrome, and Sturge-Weber syn-drome.83,84,68,133,150,151,165
EPC may be associated with widespread gliomatosis cerebri224 and paraneoplastic recurrent multifocal enceph-alitis164 and was seen in cat scratch disease.202 It can be observed in hemiconvulsion, hemiplegia, epilepsy (HHE) syn-drome.45
EPC and SPSE can occur as a complication in nonketotic hyperglycemia in children and adults, including elderly patients (>50 years).176,214,234 Such a condition may show progressive development over days. Clinical symptoms associated at the beginning are polydipsia, loss of appetite, simple partial seizures with motor symptoms, and a mild-to-moderate psycho-organic syndrome. In a hyperosmolar nonketotic coma, glucose values are often very high, and osmolarity may be >320 mmol. Because phenytoin can reinforce hyperglycemia, it is contraindicated. Renal failure giving rise to uremia in older patients is particularly often associated with frequent myoclonic jerking. In renal insufficiency, the EEG becomes frequently abnormal and is characterized by excessive slowing. Not infrequently, EEG abnormalities are enhanced during and after hemodialysis.
EPC has been described in mitochondrial disorders such as Leigh syndrome,72 mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes,9,51 subacute measles encephalopathy in immunosuppressed children, and Alpers disease with, in the case of Bourgeois and Aicardi,32 a deficiency of cytochrome c oxidase.
Iatrogenic partial motor SE has been reported after exposure to penicillin, azlocillin, or cephotaxime301 and after diagnostic use of metrizamide.229
Somatomotor SPSE occurs about 50% of the time as a first manifestation in otherwise nonepileptics, is usually seen in the elderly, and often reflects an embolic but otherwise asymptomatic ischemia (sylvian or suprasylvian). The so-called somatomotor SPSE accompanying severe brain lesions in nonepileptics presents differently, with periodically repeated, about 1/sec localized myoclonus involving, as a rule, a proximal part of limbs or one side of the trunk or flank. It is nearly exclusively observed in elderly patients, most often representing infarction in watershed territories (so-called interterritorial infarction), or, less frequently, metastatic brain tumor or brain contusion.44,166 The EEG most often shows corresponding periodic lateralized epileptiform discharges (PLEDs; Figs. 1 and 2). In vascular cases, Gastaut91 referred to the “initial myoclonic phase of interterritorial infarction” in such a situation, to avoid the term status.
PLEDs may be an ictal EEG pattern in SPSE. Ictal PLEDs are often associated with corresponding jerks in the sense of EPC. In addition, PLEDs can be associated with recurrent and prolonged episodes of a confusional state associated with psychic and neurologic manifestations. Terzano et al.248 reported seven elderly (>60 years of age) patients with PLEDs and prolonged confusional states. In these seven patients, the EEG became normal when the ictal episodes subsided, either spontaneously or following intravenous diazepam. More often, however, PLEDs are viewed as an “interictal” pattern that can last as long as 3 months to 2.5 years274 and can occur in a variety of disorders. Snodgrass et al.,238 for example, found PLEDs most often in acute unilateral lesions (vascular, 35%; mass lesion, 26%; infection, including herpes simplex encephalitis, 6%; anoxia, 2%), but also in other conditions (22%), such as alcohol withdrawal and toxic-metabolic disorders. They are also seen with chronic seizure disorders or with old static lesions when associated with recent seizures.
The determination of whether such a periodic EEG pattern, if not associated with obvious ictal signs and symptoms, reflects an interictal or ictal state (in the sense of an ongoing [“sub-clinical”] electrographic status) has important implications for antiepileptic therapy. Single photon emission computed tomography (SPECT) studies strongly suggest that PLEDs may be ictal in some patients. Whereas Treiman253 feels that treatment is appropriate until all EEG discharges are gone, Lothman et al.142 and Brenner and Schaul34 suggested that a stable periodic pattern may represent underlying pathology with neuronal damage rather than seizure activity and does not demand antiepileptic treatment.
Pathophysiology
Why and how epileptic discharges can remain restricted to a given cortical (or corticothalamic sector) region without noteworthy propagation, over long periods of time to produce SPSE might be related to (a) certain pathologies, (b) the localization of the lesion, and (c) sufficiently preserved inhibition to limit the epileptic process in space.
In general, the pathophysiology of SPSE is poorly understood. Considerable knowledge, however, has been accumulated on EPC. Bancaud’s classification of EPC into types 1 and 2 is clinically useful.22 In type 1, the myoclonias correspond to a focal structural lesion in the contralateral rolandic cortex. This type does not show any age dependence, the continuous myoclonias are restricted, and the EEG usually also shows a sharply delineated spike focus. The clinical course and the prognosis are determined by the underlying pathology. The unaffected brain regions are often and remain, as a rule, more or less normal. Type 2, also called Rasmussen syndrome,6,10,172,206 is a progressively evolving “chronic encephalitis” with a predominance in children and a peak around the sixth year of life.101,205 (see Chapter 243.) In about two thirds of cases, there is a history of a preceding nonspecific infection. Often, the disease starts with a grand mal seizure or several simple partial motor seizures and eventually includes complex partial seizures. In about 50% to 60% of patients, EPC develops and is then the predominating seizure type.172 Seizures are usually resistant to antiepileptic drugs, and extensive resective surgery was the only known effective treatment.
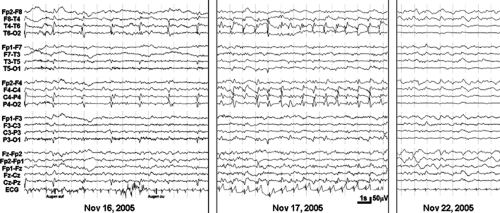 FIGURE 1. Illustration of periodic lateralized epileptiform discharges (PLEDs) followed for 1 week. On November 16, 2005 (left), there was a periodicity of sharp waves at T6 and P4 of about 0.4/s with an intermingled seizure discharge (see Fig. 2). Valproate was administered. One day later, the PLEDs showed a periodicity of 1/s, and there were now seizure discharges. Five days later, PLEDs were no longer present, but there was a flattening at T6 and P4. One day before admission (November 15, 2005), the patient experienced a left hemiplegia due to a new hyperintense magnetic resonance imaging lesion in the right crus cerebri. This was diagnosed as new plaque within the diagnosed chronic-progressive multiple sclerosis (since 1985). The patient also suffered from schizophrenia-like psychosis and anorexia nervosa. She had had several seizure-like episodes (falls during the last year but without the clear diagnosis of epilepsy). |
In relatively well-preserved brain areas, perivascular lymphocytic cuffs and glial nodules, and in later stages microcystic degeneration with marked neuronal fallout but without evidence of inflammatory elements, are typical histologic findings.208 It is fair to say that the nature of this disease remains obscure, although in recent years an autoimmune process has been postulated, as prompted by various reports.11,211,257 Autoantibodies in sera from patients with active Rasmussen
encephalitis and experimentally induced antibodies in rabbits seemed to act as agonists for glutamate receptors consisting of or containing GluR3 subunits. Agonist activity of autoantibodies on a glutamate receptor subunit suggested their role as pathogenetic factors, potentially as highly specific excitotoxins or neuromodulators. Our search, however, for the presence of anti-GluR3 antibodies in sera and cerebrospinal fluid of four patients with typical histologic findings of Rasmussen encephalitis yielded negative results.252 In other patients with Rasmussen encephalitis, autoantibodies against the glutamate receptors (GluR3, GluRepsilon2) were detected,131,211,246,247,257 suggesting an autoimmunologic etiology in a subgroup of this syndrome. Preliminary data with plasma exchange therapy in some patients with Rasmussen’s encephalitis seem also to support this etiology.221,291
encephalitis and experimentally induced antibodies in rabbits seemed to act as agonists for glutamate receptors consisting of or containing GluR3 subunits. Agonist activity of autoantibodies on a glutamate receptor subunit suggested their role as pathogenetic factors, potentially as highly specific excitotoxins or neuromodulators. Our search, however, for the presence of anti-GluR3 antibodies in sera and cerebrospinal fluid of four patients with typical histologic findings of Rasmussen encephalitis yielded negative results.252 In other patients with Rasmussen encephalitis, autoantibodies against the glutamate receptors (GluR3, GluRepsilon2) were detected,131,211,246,247,257 suggesting an autoimmunologic etiology in a subgroup of this syndrome. Preliminary data with plasma exchange therapy in some patients with Rasmussen’s encephalitis seem also to support this etiology.221,291
The definite proof for a cortical origin of EPC was provided by three studies using depth-electrode recordings (Fig. 3).21,39,287 An experimental model of EPC was achieved by injections of aluminum hydroxide into monkey motor cortex.46 Using this experimental model, Chauvel and associates proved the role of the long-loop reflexes for generation of the cortical myoclonus.47 Thermocoagulation of the thalamic nucleus ventro-lateralis posterior (VPL) oralis disrupted the reflex loop and led, in the majority of cases, to a cessation of the myoclonus.48 The participation of the (presumably) ventrolateral and intralaminar thalamic nuclei119 in the epileptic process was illustrated on fluorodeoxyglucose (FDG)-positron emission tomography (PET) by a simultaneous metabolic increase in both the cortical and the ipsilateral thalamus in a patient with an EPC.106
On the other hand, absence of epileptogenic EEG abnormalities in some patients with EPC189 and presence of subcortical brain lesions with preserved cortex31,120 led to the hypothesis of a subcortical origin of the EPC in at least some patients. A recent report even implicates a cerebellar lesion as possible cause for EPC.263
In 1985, Hallett introduced three types of epileptic myo-clonus: (a) cortical reflex myoclonus as a fragment of partial epilepsy, which represents hyperactivity of a focal area of cerebral cortex; (b) reticular reflex myoclonus as a fragment of generalized epilepsy with hyperactivity of medullary brainstem reticular formation; and (c) primary generalized epileptic myo-clonus as a fragment of primary generalized epilepsy, which may represent a generalized hyperactive response of cortex to subcortical input.107 (For further discussion of myoclonus, see Chapter 277.)
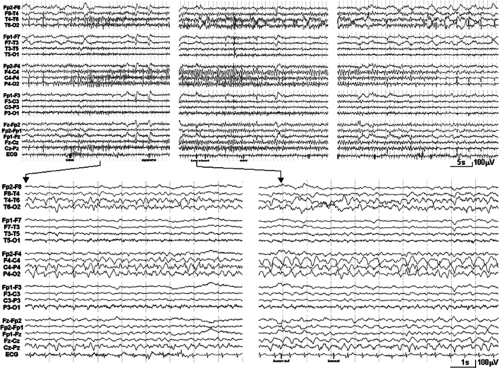 FIGURE 2. Same patient as in FIGURE 1. During the electroencephalogram (EEG) recorded on November 16, 2005, with periodic lateralized epileptiform discharges of a periodicity of sharp waves at T6 and P4 of about 0.4/s, the depicted seizure discharge occurred with evolving slow rhythmic activity at the right temporo-parieto-central region. Upper rows: Sections showing the beginning, height, and the end of the seizure. Lower row: Corresponding EEG sections (indicated) with better time resolution. |
In cases with cortical reflex myoclonus, the epileptogenic focus is localized in the contralateral rolandic cortex, and the EEG may show spikes related to the myoclonic jerks. In cases without a clear-cut temporal relation between epileptic events and myoclonus in the EEG, the back-averaging technique identifies the spikes preceding the myoclonus.227 Somatosensory-evoked potentials of the rolandic cortex are abnormally enlarged.228 Tight relations between the afferent volley evoking abnormal evoked potentials in the primary motor cortex itself and triggering of focal myoclonus have been demonstrated by depth recordings in patients with EPC.49
Without such proof of the cortical origin of the myoclonus by neurophysiologic methods, a subcortical or even spinal origin of the myoclonus has to be considered. Cockerell et al. suggested that the diagnosis of EPC should be confined exclusively to cortical myoclonus.53 They proposed the term “myoclonia continua” for myoclonus that arises extracortically.
In cases of EPC in which the jerks are associated with other seizures, the physiologic characteristics of the jerks are identical to those of cortical myoclonus.230 In this sense, cortical myoclonus is cortical epilepsy, that is, a hypersynchronous discharge from a group of cortical cells. By employing different physiologic methods, such as back-averaging of EEG recordings in relation to the jerks, evoked potentials, and electromyographic recordings of the sequence of recruitment of muscle
groups in a myoclonic jerk, a distinction of myoclonus into cortical, brainstem, and spinal myoclonus is possible in most cases. Typically in cortical myoclonus, the EEG cortical generator potentials precede the jerks; sensory-evoked potentials are enlarged; and the myoclonus may be spontaneous and action or stimulus sensitive with a rostrocaudal pattern of muscle recruitment and antagonist and agonist cocontraction. In stimulus-sensitive myoclonus, the reflex timings are compatible with a cortical loop. Obeso et al.171 reported patients showing a spectrum of spontaneous and stimulus-sensitive myoclonus, EPC, jacksonian seizures, and generalized seizures, all with similar physiology. In these cases, EPC is repetitive cortical myoclonus. In cases without seizures, however, jerks resemble EPC clinically but not neurophysiologically. In such cases the jerks might be of subcortical origin. Menini and Naquet149 called this variant type C myoclonus with suggested origin in the brainstem. It is, however, fair to say that this variety is neither as common nor as well studied as the cortical myoclonus, and its exact nosologic position is not clear.
groups in a myoclonic jerk, a distinction of myoclonus into cortical, brainstem, and spinal myoclonus is possible in most cases. Typically in cortical myoclonus, the EEG cortical generator potentials precede the jerks; sensory-evoked potentials are enlarged; and the myoclonus may be spontaneous and action or stimulus sensitive with a rostrocaudal pattern of muscle recruitment and antagonist and agonist cocontraction. In stimulus-sensitive myoclonus, the reflex timings are compatible with a cortical loop. Obeso et al.171 reported patients showing a spectrum of spontaneous and stimulus-sensitive myoclonus, EPC, jacksonian seizures, and generalized seizures, all with similar physiology. In these cases, EPC is repetitive cortical myoclonus. In cases without seizures, however, jerks resemble EPC clinically but not neurophysiologically. In such cases the jerks might be of subcortical origin. Menini and Naquet149 called this variant type C myoclonus with suggested origin in the brainstem. It is, however, fair to say that this variety is neither as common nor as well studied as the cortical myoclonus, and its exact nosologic position is not clear.
The neurophysiologic criterion for a cortical generator of myoclonus is the presence of a preceding potential in the central region contralateral to the jerk. However, many circumstances, related to the rate of occurrence of myoclonus or presence of a lesion modifying the geometry of the cortex or volume conduction, can impair the reliability of jerk-locked averaging. Applying methods that analyze EEG-electromyograph (EMG) and EMG-EMG relations in the frequency domain, several studies have demonstrated functional coupling between cortical and muscular activity during myoclonus even when back-averaging was negative.37,38,146,154 Distinction between the pathologic corticomuscular coupling and the physiologic one might be only quantitative, and polygraphic EMG recordings remain essential for identifying pathologic drives to muscles.104,261 In addition, an exaggerated coupling in the efferent pathway does not preclude the existence of putative generators upstream of the motor cortex.
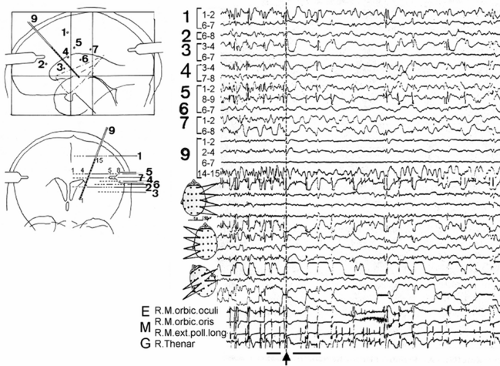 FIGURE 3. Stereo-electroencephalogram in a patient with epilepsia partialis continua recorded by eight left-hemispheric, 8-contact stereotactically inserted depth electrodes (of the “chronic” type) and by an additional rigid 15-contact electrode (#9) with its tip in the ventrolateral thalamus. Note the correspondence of left central spikes with the corresponding electromyographically (EMG) recorded muscle jerks and the different time scales (the paper speed was doubled at the time indicated by the vertical line). The patient underwent thermocoagulation of the ventrolateral thalamus with initially good, but in the long-time course moderate, therapeutic effect (follow-up >20 years; no definitive histologic diagnosis). |
Clinical Features
Ictal Signs and Symptoms
SPSE can be described from a practical standpoint within the following categories91:
Somatomotor SPSE, which, according to Passouant et al.,186 accounts for approximately two thirds of all cases of partial SE occurring in known epileptics. Somatomotor SPSE can be further divided into the following categories:
One subtype that occurs during the course or at the onset of somatomotor partial epilepsy, that is, somatomotor SPSE stricto sensu.
Epilepsia partialis continua (Kozhevnikov).
So-called somatomotor SPSE accompanying severe brain lesions in nonepileptics (“initial myo-clonic phase of interterritorial infarction with PLEDs”91).
Another special subtype, adversive SPSE, in particular, the oculoclonic form.
The less frequent sensory SPSE can be divided into the following subtypes:
SPSE with dysesthesia
SPSE with visual phenomena
SPSE with acoustic phenomena
SPSE with olfactory or gustatory phenomena
SPSE with vertigo
SPSE with autonomic phenomena, with a special subtype in children—abdominal SPSE—might be classed separately.
Dysphasic or aphasic SPSE may be divided into intermittent versus continuous and into receptive (“fluent aphasia”) versus expressive (“aphemia”) types. The Landau-Kleffner syndrome and CSWS are sometimes listed in this context. Whether this is appropriate is a matter of controversy. Arguments against it are that the core symptoms do not immediately resolve after cessation of seizures and disappearance of spikes.
French authors also have used a category erratic. Other rare manifestations of SPSE and boundary conditions can be subsumed under this category.
Finally, we have SPSE with psychic and emotional phenomena.
Somatomotor SPSE is characterized by frequently repeated typical somatomotor seizures with or without jacksonian march and with more or less pronounced EEG discharges in the rolandic region. Consciousness is well preserved, and there are no marked vegetative signs. EPC is a particular subtype with localized segmental myoclonus.
Adversive SPSE is briefly reported by a few authors43,209 but might be exceptional if we exclude cases with secondary generalization and tonic status with predominantly unilateral seizures and accompanied by head and eye turning. Oculoclonic SPSE with occipital discharges and epileptic nystagmus87,93,236 and palatal tremor168 are other rare manifestations of motoric phenomena of SPSE.
Epilepsia partialis continua is characterized by almost continuous, rhythmic muscular contractions affecting a limited part of the body for a period of hours, days, or even years. The myoclonic jerks have a frequency of about 1 to 2 per second and may persist during sleep.22,193,287 About 60% of the
patients exhibit other types of seizures in addition to an EPC,53 such as secondary generalized seizures and complex partial seizures. In addition to muscle twitching, patients may show varying degrees of muscle weakness, sensory loss, or stretch reflex changes.
patients exhibit other types of seizures in addition to an EPC,53 such as secondary generalized seizures and complex partial seizures. In addition to muscle twitching, patients may show varying degrees of muscle weakness, sensory loss, or stretch reflex changes.
The presentation of the disorder depends on the underlying cause (see section Etiology). Patients with localized neoplastic, vascular, or infectious brain lesions may have neurologic deficits and isolated seizures prior to the onset of the focal SE.19 With metabolic causes, however, such as nonketotic hyperglycemic diabetes mellitus, or hypersensitive reactions to certain drugs such as penicillin or metrizamide, the onset of focal SE is sudden.219
In the Russian patients, the epilepsy usually developed 3 to 4 months after a febrile illness, associated with a hemiplegia or monoplegia in 30% of cases. In this condition, the jerks typically affect agonist and antagonist muscles with a rhythmic quality, in short bursts of 1 to 2 seconds’ duration alternating with quiescent phases 2 to 4 seconds long, persisting in sleep and worsened by action or stress. Motor seizures with jacksonian march or generalized epileptic seizures are almost invariable accompaniments, although they have a strong tendency to improve over time. The jerking, often highly focal, continues relentlessly for years. Sensory symptoms occur in about one fifth of cases, and 80% have a persisting hemiparesis. EPC associated with Rasmussen encephalitis manifests itself in children in the majority of cases (mean age 6.8 years, with 85% being <10 years of age). All children present with epileptic seizures, often generalized tonic–clonic, although focal simple or complex partial seizures also occur as a first manifestation of the disease. In Rasmussen encephalitis, about one half of the patients exhibit episodes of EPC, usually within 3 years of onset with epilepsy. These episodes last hours to years and are often discontinuous. The condition is progressive, and after a highly variable period of 3 months to 10 years, fixed focal deficits develop, notably hemiplegia, hemianopia, and (depending on the hemisphere) aphasia, as well as progressive intellectual impairment. After an initial progressive course in which the focal motor seizure activity is often multifocal, the disease process appears to eventually burn itself out, at least in a substantial proportion of Rasmussen encephalitis patients.162
With regard to localization, EPC is a particular form of rolandic partial epilepsy that involves the motor strip of one hemisphere and usually has a clinically localized appearance. In Rasmussen encephalitis, the disease process seems to be more widespread, with diffuse patchy inflammatory changes in the cortex and white matter (microglial nodules, perivascular cuffs of small lymphocytes and monocytes, multifocal neuronal loss, and some spongy degeneration) depending on the features of the disease activity. (See also Robitaille,208 who classified the Montreal specimens into four groups of disease activity.)
Current views are that the physiologic characteristics of the jerks in most cases of EPC are identical to those of cortical myoclonus (see section Pathophysiology). The myoclonic jerks in EPC can affect any muscle group. They may be confined to a single muscle or muscle group or they may be widespread. Face, upper limb, and trunk predominate. The distribution of jerks can vary over time. Agonists and antagonists are affected together, and distal muscles are affected more often than proximal ones. Isolated clonic twitching of the abdominal muscles due to a metastatic cortical lesion and considered as a rare manifestation of EPC has been described.78 Jerks are unilateral. Bilateral cases have been included,141,249 but it is questionable whether such cases should be described within the category called EPC. Takahashi et al.245 studied EPC of childhood involving bilateral brain hemispheres; Ashkenazi et al.16 described a bilateral focal motor status epilepticus with retained consciousness after stroke; and Lim et al.140 described a generalized myoclonus evolving into EPC due to a cingulate gyrus venous angioma.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


