CHAPTER 216 Split Spinal Cord
Descriptions of split cord malformation (SCM) appeared in the medical literature as early as the 17th century.1,2 Historically, the terms diastematomyelia (Greek for “cleft[ed] medulla”) and diplomyelia (Greek for “double[ed] medulla”) were used to describe developmental anomalies now grouped together as split cord malformation.3 The sine qua non of these developmental anomalies is the presence of a longitudinal cleft within the spinal cord across one or more vertebral segments.
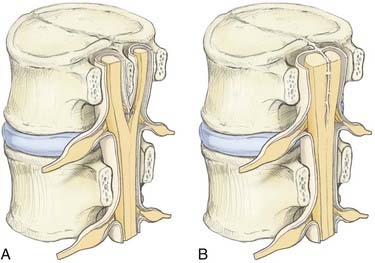
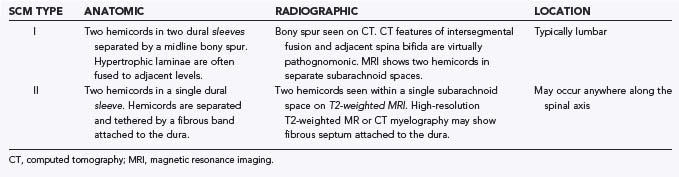
Type I malformations (formerly diastematomyelia) are characterized by a bony septum that cleaves the spinal canal in the sagittal plane and a duplicated thecal sac (Fig. 216-1A).3 Type II malformations (formerly diplomyelia) are characterized by a cleft cord within a single dural sac, often tethered by a fibrous midline septum to the adjacent dura (Fig. 216-1B and Table 216-1). Not all SCMs fit these descriptions precisely, but the classification scheme provides a framework for the surgical approach to these conditions. In 1992, Pang and colleagues proposed a unified theory attributing both type I and type II SCM to a single error in embryogenesis: adhesion between the ectoderm and endoderm leading to a persistent neurenteric canal.3
Not uncommonly, SCMs are accompanied by nonfunctional paramedian nerve roots traveling within a fibrovascular bundle to or beyond the dura. A fibrous tract extending from the epidural space to a small overlying plaque of atretic skin may also be present. Both the intradural bands and the associated skin lesion have been referred to by the term myelomeningocele manqué, which is derived from the French verb manquer (“to miss”) and reflects the outmoded theory that myelomeningocele manqué is a forme fruste of true myelomeningocele.4 Instead, the descriptive nomenclature “dorsal bands” is used here.
Epidemiology of Split Cord Malformations
Although their precise incidence is unknown, SCMs are exceedingly rare and represent 3.8% to 5% of all congenital spinal anomalies.3,5 One recent series estimated the prevalence of SCM to be 1 in 5000 (0.02%) live births.6 Similar to other forms of occult spinal dysraphism, there is a slight female preponderance, approximately 1.3:1.7–10 The peak age at initial medical evaluation is 4 to 7 years, although there is a second peak between 12 and 16 years of age coinciding with the postpubescent growth spurt. A delay in diagnosis until adulthood may be seen, with back pain being the sole complaint7,8,10,11 or pain accompanied by progressive neurological deficits that may be mistaken for degenerative disease. Although estimates of prevalence vary, type I SCMs may occur more frequently than type II lesions.8,10,11
SCMs rarely occur in the absence of associated anomalies. The most common associations (in descending order) are a tethered/low-lying cord (>50%), kyphoscoliosis (44% to 60%), syringomyelia (27.5% to 44%), and spina bifida (11% to 26%).7,8,10 Conversely, SCMs may occur in 5% of patients with congenital scoliosis12 and in up to 15% of patients with myelomeningocele.13,14 When SCM is seen in association with myelomeningocele, it is three to four times more likely to be a type I malformation and tends to occur at or just rostral to the level of the myelomeningocele defect. On occasion, multiple dysraphic lesions may be identified, and occult SCMs can account both for a discrepancy between functional level and anatomic level of the myelomeningocele placode and for late neurological deterioration in patients with myelomeningocele and an undisclosed SCM.11 Dermoids, dermal sinus tracts, and neurenteric cysts may also be found in conjunction with SCMs.
Embryogenesis
SCMs are thought to occur during the third week of embryogenesis, concurrent with gastrulation, as a result of failure of fusion of the notochord in the midline and subsequent persistence of an accessory neurenteric canal.1,3,15,16
On day 17 of human development, cells of Hensen’s node ingress at the embryonic midline to begin formation of the notochord. In a process of cell-to-cell intercalation, bilayers of cells ingress from each side of midline and intermix to form a singular midline notochordal process.17 Oriented cellular division and cell-to-cell intercalation contribute equally to the process of convergent extension by which the notochord elongates. Oriented cellular division refers to the preferential direction of mitosis demonstrated during embryogenesis that allows a mass of cells to extend in a particular plane rather than growing spherically. Intercalation refers to the cellular process by which two groups of cells integrate in a mosaic pattern (Fig. 216-2A). Thus, polar integrated growth of the combined mass occurs in the cranial-caudal direction. Cellular intercalation is partially governed by genes of the wnt family, which are also responsible (along with sonic hedgehog) for dorsal-ventral patterning of the vertebrate nervous system.18 Knockouts of Prickle, another cell polarity molecule that is conserved across species, demonstrate a foreshortened and thickened notochord.
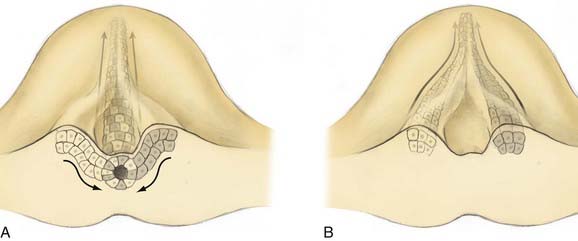
Reciprocal induction of notochordal and paraxial mesodermal cells also plays a key role in this process. Mesodermal somites express protocadherin, which promotes midline intercalation of the notochord. Embryos lacking protocadherin demonstrate normal elongation of the paramedian notochord masses but no integration, reminiscent of the failed integration seen over small spinal segments in human SCM19 (Fig. 216-2B). Once formed, the notochord acts as an organizer by secreting multiple morphogens that are responsible for proper neurulation of the embryo. Experimental transplantation of notochord fragments has demonstrated that presence of the notochord is necessary and sufficient to influence patterning of spinal development20 and that ectopic or redundant notochordal tissue may result in complete duplication of the spinal cord and surrounding tissues.21 Emura and colleagues produced an amphibian model of SCM by microsurgically creating a neurenteric fistula in embryos, which were then incubated to term. They observed SCM in 40% of the embryos that survived the procedure.22
Most theories posit two common prerequisites for the development of SCM.1,3,23,24 The first is persistent adhesion of the ectoderm and endoderm, termed an accessory neurenteric canal by Bremer.24 The second is a cleft notochord, with subsequent induction of a cleft spinal cord. In 1992, Pang proposed a unified theory for the development of both types of SCM in which these two prerequisites occur as the result of a single ontogenic event—failure of midline integration of the anlagen destined to become the notochordal process.3 This theory postulates that the mass of cells derived from the primitive pit does not ingress as a single mass but instead exhibits left-right asymmetry whereby it courses mediolaterally from the primitive pit before reintegrating in the midline (Fig. 216-2B). Failure of reintegration leads to the formation of an accessory neurenteric canal, which Pang labeled the endomesenchymal tract.11 Persistence of the endomesenchymal tract results in a median cleft with two heminotochords, which induces a similarly cleft spinal cord.
This unified theory explains the subsequent occurrence of types I and II SCM based on incorporation of the meninx primitiva into the endomesenchymal tract. Inclusion of meninx primitiva cells, which arise around day 30, results in the formation of two completely separate dural sleeves as seen in a type I SCM. Inasmuch as the meninx primitiva is osteogenic, this also results in the formation of a midline bony spur (see Fig. 216-1A). When the meninx primitiva is not incorporated into the endomesenchymal tract, a single dural sleeve (and single arachnoid space) contains two hemicords separated by a thin fibrous band, as seen in a type II SCM (see Fig. 216-1B). Incorporation of neural crest (other than the meninx primitiva) into the endomesenchymal tract results in the formation of neural elements medial and dorsal to the hemicords that extend to the extradural space and result in the formation of dorsal bands.
The presence of two separate notochordal anlagen during formation of the notochordal process has never been directly observed, but knockout mice lacking genes for intercalation develop two heminotochords that are not fused in the midline.19 The unified theory suggests that caudal failures of midline integration, which occur relatively late during embryonic development, are more likely to coincide with emergence of the meninx primitiva and thus produce type I SCMs. In fact, type I SCM most frequently occurs from L2 to L4 and is rarely seen above the thoracolumbar junction, whereas type II SCM occurs almost exclusively above T8.8
Signs and Symptoms
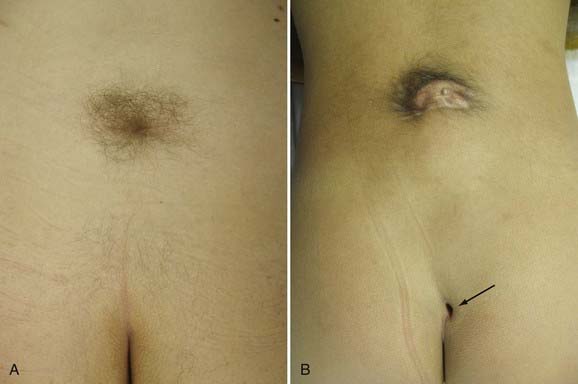
Patients with split cord may be asymptomatic, with only cutaneous stigmata of their spinal dysraphism. In various series, there is some type of cutaneous mark present in up to 92% of patients.7,10,11,14,25 The common stigmata associated with SCM are summarized in Table 216-2.7–11,14,25,26 Because of the rostral migration of neural elements relative to the bony spinal column that normally occurs after the formation of an SCM, cutaneous markers are found somewhat caudal to the level of the neural defect itself. The presence of hypertrichosis, although variable, is highly specific for SCM (Fig. 216-3A). One report identified an odds ratio of 61.6:1 in favor of the occurrence of an SCM if a hairy patch is present.25 One characteristic variation of hypertrichosis, the “faun’s tail,” consists of a patch of unusually coarse, raised hair that is strongly associated with type I SCM.25 There is typically a capillary hemangioma underlying these hairy patches. On occasion (≈7%), hemangiomas alone, without overlying hypertrichosis, may indicate the presence of an SCM. Although sacral dimples, dermal sinuses, and cutaneous lipomas are not specific to SCM per se, they often indicate the presence of an additional underlying dysraphic lesion in a patient with SCM (Fig. 216-3B). True sacral dimples that are suggestive of underlying dysraphism should be distinguished from shallow, symmetrical dimples over the coccygeal midline. The latter is common in the general population and is not indicative of the presence of distal tethering pathology.
TABLE 216-2 Clinical Signs and Symptoms
| SYMPTOMS | PREVALENCE (%) |
|---|---|
| Backache11,26 | 30-50 |
| Progressive neurological deficit11 | 66 |
| Urinary retention/incontinence10,26 | 13-32.5 |
| Leg pain/hyperesthesia10,26 | 7-10 |
| Asymptomatic10 | 13 |
| CUTANEOUS MARKERS | |
| Hypertrichosis9–1125 | 31-61 |
| Dermal sinus8–10 | 7-40 |
| Subcutaneous lipoma8–1014 | 2-25 |
| Sacral dimple8,10 | 5-10 |
| Capillary hemangioma8–1014 | 0.5-7.2 |
| ASSOCIATED FINDINGS ON EXAMINATION | |
| Abnormal urodynamics26 | 74 |
| Left-right motor/sensory asymmetry8 | 21 |
| Left-right asymmetry (limb size/length)10 | 12-15 |
| Talipes equinovarus7,10 | 17-20 |
A majority of patients with SCM have one or more signs and symptoms of neurological deterioration.11 Axial back pain is common and is typically focal and intense around the levels of the split cord. Adults with undiagnosed SCM may have back pain as the only symptom. A radicular component may be present in 10% of both adult and pediatric patients.10,25
Progressive sensory or motor dysfunction (or both) is seen in two thirds of patients. There are two separate, but interrelated causes of asymmetrical motor findings in the lower extremities. Nearly 50% of patients have gross (i.e., structural) asymmetry of the lower extremities, a condition referred to as neuro-orthopedic syndrome. This syndrome is characterized by a triad of limb length discrepancy, muscular atrophy (resulting in secondary weakness), and clubfoot deformity (talipes equinovarus).27–29 The smaller limb is often ipsilateral to a smaller hemicord. Fewer patients (about ≈20%) have an asymmetrical neurological deficit despite structural symmetry of the limbs. This finding is highly associated with hydromyelia of one hemicord.8
Autonomic dysfunction is typically manifested as urologic signs and symptoms. Subjective complaints of enuresis are a poor indicator in comparison to objective urodynamic testing. Although only about one third of patients typically have complaints of urinary dysfunction, urodynamic studies demonstrate abnormal bladder function in nearly three quarters.26 Detailed urologic assessment should be undertaken as part of the initial evaluation and before any surgery.
Surgeons evaluating a patient for suspected SCM should be cognizant of associated anomalies that may require additional surgical intervention. Summarized in Table 216-3,7,8,10 associated anomalies include scoliosis, tethered cord secondary to a thickened filum or terminal lipoma, myelomeningocele (with associated Chiari II malformation), and syringomyelia.
TABLE 216-3 Associated Anomalies
| ANOMALY | PREVALENCE (%) |
|---|---|
| Scoliosis | 44-60 |
| Thickened filum terminale | 22-71 |
| Neuro-orthopedic syndrome | 20-56 |
| Syrinx | 28-44 |
| Myelomeningocele | 11-26 |
| Terminal lipoma | 9-16 |
| Chiari malformation | 6-7 |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


