8 Surgical Treatment of Craniopharyngiomas Craniopharyngiomas are tumors usually found in the region of the infundibulum, although they can develop anywhere along an axis from the nasophayrnx to the third ventricle. They comprise ~5 to 10% of pediatric brain tumors and present with signs and symptoms referable to their location, including visual loss, hormonal disturbances, hydrocephalus, and headache. The broad spectrum of management strategies employed in the treatment of these tumors—including surgical resection, radiation, and intratumoral delivery of chemotherapeutic agents or radioisotopes—underscores the difficulty of achieving successful cures with acceptable morbidity and has fostered considerable controversy among physicians involved in the care of children with these lesions. This chapter reviews the etiology, pathology, epidemiology, and presentation of these tumors, outlines relevant initial diagnostic evaluations, and summarizes the surgical management of craniopharyngioma. The term craniopharyngioma was coined by Harvey Cushing in 1932 to describe a class of tumors found in the sellar region that were first reported by Erdheim in 1904.1,2 Microscopic examination has revealed that craniopharyngiomas comprise two distinct subtypes, adamantinomatous and papillary, which some consider to have separate—if related—embryologic origins (mixed subtypes of tumor, with both adamantinomatous and papillary regions, have also been described).3–5 Evidence supports the premise that craniopharyngiomas originate from cells derived from the development of the adenohypophysis and tend to arise primarily from the region of the infundibulum.6,7 At the end of the first month of gestation, part of the oral cavity (the stomodeum) projects up toward the brain (Rathke pouch). This tissue interfaces with the infundibulum: a downward projection of the brain. Over the next 2 weeks, the connection between the oral cavity and the brain (the pharyngohypophyseal stalk within the craniopharyngeal duct) involutes as the sphenoid bone grows and closes off the two spaces, while Rathke’s pouch contributes to the development of the anterior pituitary gland.6,6–8 Craniopharyngiomas are thought to arise from remnants of Rathke’s pouch and tissue from the craniopharyngeal duct.9,10 In particular, there is some evidence suggesting that adamantinomatous lesions are derived from neoplastic transformation of epithelial remnants of the craniopharyngeal duct.8,9 This is in contrast to the squamous papillary subtype of craniopharyngioma, which is considered to result from the metaplasia of adenohypophyseal cells.8,11 These tumor subtypes differ pathologically in several ways. Adamantinomatous craniopharyngiomas are most common in children and share characteristics with tooth enamel–forming and long-bone tumors of the skeletal system (the eponymous adamantinomas), including the tendency to produce calcified deposits intratumorally. The epithelial cells have a keratinized squamous layer that flakes offand degenerates into a characteristic cholesterolrich “crank-case oil” fluid.4,10,11 In contrast, the papillary subtype (also called squamous papillary), found nearly exclusively in adults (albeit still less frequently than adamantinomatous tumors), is characterized by stratified squamous epithelium that does not usually exhibit the calcification or cystic degeneration evident in the adamantinomatous tumors.4,10–12 Overall, most reports indicate that more than 90% of all pediatric craniopharyngiomas are adamantinomatous; fewer than 2% are purely papillary and the remainder exhibit mixed features.10–13 In adults, approximately two-thirds (66%) of all cases are adamantinomatous, about one-fourth (27–28%) are papillary, and the remainder are mixed.10–13 Recent efforts in molecular biology have made inroads toward characterizing biological pathways involved in the genesis of these tumors. Disruptions in apoptotic pathways involving β-catenin and Wnt may contribute to the neoplastic transformation of craniopharyngiomas.8 Of particular interest is the role that growth factors and angiogenic peptides such as cathepsins (proteinases that regulate tumor invasiveness and end-effector proteinases such as matrix metalloproteinases [MMPs]) and vascular endothelial growth factor (VEGF) play in craniopharyngioma growth and development.8 These investigations are relevant not only because they shed light on the basic mechanisms of tumorigenesis but also because they facilitate the development of novel methods of tumor detection and follow-up; for example, recent data suggest that urine testing may be able to noninvasively detect brain tumors, including craniopharyngiomas.14,15 In the United States, ~340 craniopharyngiomas are diagnosed annually in the combined pediatric and adult population, of which 100 are in children between 0 and 14 years of age.16 Craniopharyngioma has an incidence of 1.3 cases per million person-years, with no significant difference in presentation by sex or race.16 As noted, about one-third of cases occur in children, and the remaining two-thirds present in adults.1,16 This distribution manifests itself in a bimodal pattern, with the largest peak at 5 to 14 years of age and a smaller one at 50 to 74 years of age.1,16 However, craniopharyngiomas have been diagnosed prenatally and in patients older than 70 years of age.17–22 Craniopharyngiomas represent 5 to 10% of all intracranial tumors in children in many series and more than half of all sellar region tumors in this population.11,16,23 They are the most common nonglial intracranial tumors in children.24 These tumors are comparatively less common in adults, comprising fewer than 5% of all brain tumors in this age group (with many reports indicating numbers closer to 1%).16,23 Although craniopharyngiomas can be discovered in the asymptomatic patient, the vast majority of cases are identified following the onset of characteristic signs and symptoms.24 These are directly related to the location of the tumor, and variations in the site of origin help to explain variations in presentation. Generally, craniopharyngiomas exhibit one of three growth patterns: prechiasmatic (affecting the optic nerves and chiasm), retrochiasmatic (affecting the hypothalamus, optic tracts, and drainage of cerebrospinal fluid [CSF]), and sellar (affecting hormonal function). Overall, the most common clinical findings include sequelae of increased intracranial pressure due to mass effect and hydrocephalus (especially headache, nausea, and vomiting); visual loss; and/or endocrinologic dysfunction.6,20,24 In the published series from Children’s Hospital in Boston, findings resulting from increased intracranial pressure were the most common, leading to discovery of the tumor in 44% of patients.24 Nearly one-fourth of patients with craniopharyngioma have hydrocephalus at presentation.20 Visual deterioration can be quite severe before detection by the family or clinicians and may be asymmetric, depending on the growth pattern of the tumor.24,25 Careful documentation of visual function is critical because it may substantially influence subsequent planning for operative approaches. Similarly insidious is the development of endocrine dysfunction, which may remain unnoticed for long periods of time owing to the often subtle onset of symptoms such as growth delay. Although a deficiency of growth hormone is the most common endocrinopathy found in the setting of craniopharyngioma (followed by hypothyroidism and diabetes insipidus), abnormalities of any and all of the pituitary hormones may be manifested, and careful retrospective analysis reveals some form of endocrine dysfunction in 60 to 90% of patients at diagnosis.24,26 Hormonal symptoms can be compounded by effects of hypothalamic injury, including temperature intolerance and dysregulation, weight gain, and behavioral disturbances. Recognition of the signs and symptoms described above prompts imaging studies of the brain to identify proximate causes. In the United States, computed tomography (CT) and magnetic resonance imaging (MRI) are commonly obtained. Tumors are usually large enough to be readily identified on standard studies, but in rare cases, smaller lesions may be missed if not specifically targeted. Multiplanar reconstructions and thin cuts through the region of the sella and infundibulum may help to reveal subcentimeter masses. MRI is particularly useful for the identification of tumors and delineation of the relationship of the tumor to surrounding neurovascular structures (including the carotids and their branches), optic apparatus, pituitary gland and stalk, and hypothalamus/ventricular system (Fig. 8.1). The tumor may appear heterogeneous, with cystic components bright on T1 and T2 and solid portions of the tumor exhibiting variable enhancement following administration of contrast (Fig. 8.2). MRI with axial, sagittal, and coronal planes with and without contrast is critical to preoperative planning and postoperative follow-up. Increasingly, MR angiography is helpful in delineating vascular anatomy for these same reasons. We feel that CT is important in diagnosing and planning surgery for craniopharyngiomas. A distinguishing feature of these tumors is the presence of calcium, which may be difficult to detect on MRI (Fig. 8.3). Regions of calcification are present in the majority of pediatric tumors (up to 90%) and more than half of adult lesions.10,23,27 Preoperative radiographic visualization of solid calcium deposits (when present) is invaluable to the surgeon in determining the feasibility of specific operative approaches. Moreover, CT is helpful in ascertaining the degree of pneumatization of the sphenoid, ethmoid, and frontal sinuses—relevant to transsphenoidal approaches for sellar tumors (sphenoid), drilling down the planum sphenoidale for frontal approaches (sphenoid/ethmoids), and bifrontal approaches for suprasellar tumors (frontal). Preoperative angiography is generally not helpful in the evaluation of craniopharyngioma. The tumors are usually fed by small vessels that are difficult to visualize, even with a dedicated angiogram, and there is no role for preoperative embolization. Most of the relevant structural anatomy can be better delineated with MR angiography, which allows visualization of the parenchyma and tumor side by side with associated vessels. The differential diagnosis of tumors in the hypothalamic/sellar region is large and includes congenital lesions (Rathke cleft cyst, arachnoid cyst, dermoid/epidermoid cyst, hypothalamic hamartoma); tumors (pituitary adenomas, germinomas/nongerminomatous germ cell tumors, lymphoma, meningiomas, schwannomas of the cranial nerves, optic gliomas); vascular lesions (aneurysm, cavernous malformation); and inflammatory conditions (neurosarcoid, lymphocytic hypophysitis).9,10,28 The characteristic calcification and cystic regions of craniopharyngiomas (especially in children) often help substantially with confirming the radiographic diagnosis. At our institution, we routinely obtain both CT and MRI in patients with suspected craniopharyngiomas. In addition to the previously noted imaging, patients with craniopharyngioma should undergo preoperative endocrinologic assessment.1,24,29 It is our practice to consult the endocrinology service before planned surgery and to obtain a panel of laboratory studies to evaluate pituitary function (Table 8.1). Deficient hormones are replaced as needed, with particular attention to cortisol deficiency: patients with craniopharyngioma should be considered in need of supplemental stress-dose steroids perioperatively. When possible, a formal assessment of visual fields and an ophthalmologic examination should be performed before surgery.24,25 This not only establishes a baseline, but also may help to guide the surgical approach if one optic nerve is substantially impaired and the other has retained function. Careful investigation and documentation of the patient’s neurologic status is important for similar reasons. Lastly, although often not practical in the setting of an ill patient, those who present in a more elective fashion may be candidates for a detailed neuropsychological evaluation, allowing caregivers and families to more effectively follow changes over the course of treatment and develop more tailored coping strategies.30,31 For those patients who present when acutely ill, two conditions should be considered. First, hydrocephalus is a common cause for rapid deterioration in the setting of craniopharyngioma. If present, placement of an external ventricular drain can often immediately decrease intracranial pressure so that the patient can be stabilized until a definitive surgical treatment can be performed. On rare occasion, a large cyst within the tumor can be drained in this fashion to reduce mass effect. Second, as mentioned previously, many patients with craniopharyngioma are cortisol-deficient, and profound deterioration can occur from apparently minor physiologic stressors. Replacement of corticosteroids frequently averts disaster in these cases (we commonly administer dexamethasone 1–4 mg intravenously, although hydrocortisone 30 mg/m2 is also effective). Once identified, the primary objective of craniopharyngioma treatment is the restoration and preservation of neurologic function with minimal morbidity. There is substantial debate regarding the most effective method for achieving this goal. The difficulty in developing definitive guidelines for care stems in large part from the variability in presentation, the need for exceptionally long follow-up to assess efficacy, and the rarity of these tumors. The preferential use of specific treatments—radical surgery, subtotal resection, radiation, intracystic administration of chemotherapy—may be influenced by both published data and institutional bias. Here, we present an overview of surgical techniques used in the management of craniopharyngioma—approaches that are of use for both radical and subtotal resections. Before an operation is undertaken, it is critical to define the goals of surgery. An important distinction is whether the objective is a complete resection or a planned subtotal debulking. This topic is controversial, and data exist supporting both strategies.1,6,24,32–37 The policy at our institution is to attempt a total resection at initial presentation in most cases, but exceptions are made when preoperative imaging suggests that surgical morbidity would be unacceptable.24 Radical resection can be attempted on repeat operations, but our experience has been that most successful reoperations occur when tumor was left behind initially because of poor visualization—a problem that can be remedied by altering the approach. In contrast, patients with tumors that were inextricably attached to vital neurovascular structures at the first operation are often poor candidates for gross total resection the second time around. Formulating goals for the surgery and planning an operative approach are difficult, and data support that surgeons with greater experience achieve better outcomes.6,32,34 We routinely discuss cases among our group to draw upon the collective expertise of our practice. Unfortunately, even with careful planning, the nature of these tumors sometimes precludes achieving the expected preoperative objectives. The primary goal of surgery is resection of the lesion with preservation of vital neural and vascular structures. Factors such as anatomic constraints imposed by these vital structures or characteristics of the tumor (eg, areas of calcification) may preclude a gross total resection, and secondary goals of surgery may include reducing mass effect, debulking the tumor so that it becomes more amenable to radiation therapy (either by reducing the size or creating margins around vital structures to minimize dose effect), restoring patterns of CSF flow, and establishing a tissue diagnosis with pathologic specimens. Review of the preoperative radiographic studies is invaluable in the selection of a surgical approach best suited to achieve these goals. Variation in the size and location of craniopharyngiomas, from small lesions located solely in the sella to huge tumors filling the third ventricle, has resulted in the development of numerous distinct surgical approaches. For many surgeons, familiarity with one method will often prompt its use in the majority of cases, and it can be difficult to balance the theoretic advantages of an alternative approach against the benefit afforded by experience. It is important to understand that there can be several equally valid approaches that may be efficacious for a given tumor. The growth pattern of an individual tumor can direct a surgeon to a particular approach. In general, craniopharyngiomas fall into three distinct groups: suprasellar/prechiasmatic, suprasellar/post-chiasmatic, and sellar. When viewed through this lens, it can be helpful to pair the common approaches (subfrontal, pterional, subtemporal, transcallosal/transventricular, and transsphenoidal) with specific growth patterns to maximize access to the tumor (Table 8.2). Review of the case with nursing and anesthetic staffwell beforehand allows anticipation of needed medications, hormone replacement, blood products, equipment, and potential emergencies. In addition to a discussion of the case with colleagues, a candid assessment of expected outcomes and risks of the surgery with the patient and family is vital. In the operating room, needs such as abdominal fat grafts, pericranial flaps to provide vascularized coverage of defects, and placement of ventricular or lumbar drains are best considered well in advance of the initial preparation. In planning operative approaches, it is useful to allow for additional exposure (through extension of the incision and bone flap) if possible. Although often not needed, the ability to improve access to the tumor intraoperatively can be invaluable. The subfrontal approach is commonly used in our institution. It allows excellent visualization of the optic nerves, carotids, and lamina terminalis. This approach is useful for most craniopharyngiomas, albeit less so for isolated sellar lesions. It affords wide access to the suprasellar region and can easily be combined with other approaches, such as the pterional, transcallosal/intraventricular, and even sellar (with drilling of the planum sphenoidale). The approach can be unilateral or bilateral, depending on the anatomy of the tumor. For retrochiasmatic and intraventricular tumors, removal of the superior orbital rim and roof improves visualization while minimizing retraction on the frontal lobes (Fig. 8.4). Limitations of the approach include the risk for anosmia with injury to the olfactory nerves, the concern of venous congestion resulting from ligation of the superior sagittal sinus (which is usually minimal), and the extensive nature of the craniotomy; including violation of the frontal sinus with the attendant risk for CSF leak and mucocele. In patients with hydrocephalus, placement of a preoperative drain may relax the brain and facilitate visualization with minimal retraction. A bicoronal incision is then marked out, with preparation of the surgical field to include the possibility of a pterional extension if needed. In patients with a large frontal air sinus, a site for an abdominal fat graft is readied. Anesthesia can be forewarned about the possibility of blood loss and air embolus if a bilateral exposure is planned and the sagittal sinus is to be exposed. The patient is pinned in extension with the head midline to allow the frontal lobes to fall away from the floor of the anterior fossa. Following incision, the pericranium is preserved in elevating the scalp flap in anticipation of the need of a vascularized pedicle to cover the frontal sinus. In a unilateral case, the operating table can be rotated to vary lines of sight while the surgeon remains in a comfortable operating position, seated or standing. Dural opening should be low to the floor of the anterior fossa, and in the case of bilateral exposure, care should be taken to preserve at least one olfactory nerve. If the orbital rim and roof are removed, the surgeon should attempt to avoid injury to the periorbita because the retraction of fat can be troublesome and can result in significant postoperative bruising. Gentle retraction on the orbit can greatly enhance visualization of the tumor, although some patients may experience bradycardia with compression of the eye. This will often resolve with slight repositioning of the retractor. In retrochiasmatic tumors, opening of the lamina terminalis can be useful. Care must be taken to avoid blind traction or injury to the walls of the third ventricle because hypothalamic injury can be devastating. Gentle downward pressure on the tumor from the lamina terminalis can deliver lesion into the more accessible subchiasmatic space. Use of a dental mirror or angled endoscope can substantially aid in visualization of difficult regions of the operative field. Drilling the planum sphenoidale can increase working room and afford greater access to the sella. When this is done, attention must be given to the possibility of entering the ethmoid or sphenoid sinus and appropriate steps taken to prevent CSF leak if a breach occurs (namely, packing the sinus with fat or muscle and covering the defect with tissue, preferably vascularized pericranium). The pterional approach is often used with suprasellar tumors that are both prechiasmatic and retrochiasmatic. It offers a more lateral view than the subfrontal route and can be combined with other approaches (subfrontal, subtemporal, transcallosal/transventricular) to expand access to larger masses. In addition to this versatility, it has the advantage of being familiar to many neurosurgeons. The primary limitation of the pterional approach is reduced visualization of the contralateral opticocarotid complex (Fig. 8.5). At the Children’s Hospital in Boston, we often employ this approach for small (usually incidentally discovered) tumors that are suprasellar. Our preference is to perform a right-sided craniotomy (assuming this to be the nondominant hemisphere), although we alter this based on tumor anatomy and function of the optic nerves. Should one optic nerve have markedly impaired function, we commonly approach from the side of the healthy nerve to better visualize and protect it. The patient is positioned supine with the head extended and turned ~30 degrees away from midline. As with the subfrontal approach, the use of ventricular drainage may be helpful, although is it less likely to be needed with smaller tumors. Aspiration of CSF following the opening of CSF cisterns will nearly always enable excellent brain relaxation (again, without the need for the administration of hyperosmolar agents). Depending on the size of the tumor and the preference of the surgeon, the sylvian fissure can be opened to increase exposure at the bifurcation of the carotid. The subtemporal approach is employed primarily for access to suprasellar retrochiasmatic craniopharyngiomas that extend down toward the pons or laterally under the temporal lobe. The patient is placed in a supine or partially lateral position with the nose parallel to the floor. The vertex is tilted down toward the floor to allow the temporal lobe to fall away from the middle fossa. Upon opening the bone flap, it is helpful to use a drill or rongeur to remove bone flush with the floor of the middle fossa. Care must be taken to seal mastoid air cells with bone wax to prevent CSF leak postoperatively. Following gentle retraction on the temporal lobe, the tentorium can be divided to increase visualization of the interpeduncular and prepontine cisterns. It is important to preserve the trochlear nerve by cutting the tentorium behind the point at which they cross. Generally, the membrane of Liliequist will provide a barrier between the tumor and the basilar artery, although this is not an absolute constant, especially in reoperations. Transcallosal/transventricular approaches are most helpful for tumors located high within the third or lateral ventricles. This route can be used independently or in combination with other approaches. The main limitation is the inability to visualize neurovascular structures near the sella. Generally, a craniotomy is made over the superior sagittal sinus with one-third posterior to the coronal suture, two-thirds anterior, and extension about 4 cm off midline toward the desired side. The white appearance of the corpus callosum must be identified, and frameless stereotaxy can be helpful in determining an entry point. A 2-cm opening in the callosum is made, and one can either carefully dissect between the fornices to enter the third ventricle directly or enter the frontal horn of the lateral ventricle, then open from the foramen of Monro posteriorly along the choroidal fissure. Care must be taken to avoid injury to the fornices and internal cerebral veins. The transsphenoidal approach has generally been employed in the removal of sellar craniopharyngiomas. Although it was traditionally limited to purely sellar lesions, surgeons have become increasingly capable of successfully resecting sellar lesions with suprasellar extension, with the improved visualization afforded by the operating endoscope. The main advantages of the transsphenoidal approach (either microscopic or endoscopic) include decreased surgical morbidity, shorter hospital stays, and improved access to sellar tumors. However, transsphenoidal surgery has markedly limited lateral exposure, minimal ability to control bleeding, and an increased risk for CSF leak/meningitis. Rare but catastrophic injury can occur following injuries to the carotid arteries and their major branches. Moreover, heavily calcified lesions are difficult to remove with the access provided from this route. Low tumors (particularly ones that are primarily midline, infradiaphragmatic, and cystic) are well suited to this approach. Complication avoidance includes careful preselection of appropriate cases, as described above. In young children, the small working spaces and nonpneumatized sphenoid do not preclude this approach, but do require consideration. With children or in adults with small nares that might prevent the easy passage of surgical instruments, the translabial/transsphenoidal approach can be used to afford greater visualization and working access (Fig. 8.6). Image guidance, with either frameless stereotaxy and/or fluoroscopy, has proved useful in our institution (and others) to maintain a safe midline approach, especially in reoperated cases. On occasion, the decision to stage a procedure and perform a craniotomy at a subsequent date to remove suprasellar portions of a tumor may be prudent. Following tumor removal, careful inspection of the cavity with an angled (30- or 70-degree) endoscope should be performed to assess the completeness of the resection and ensure the absence of injury. We routinely use a periumbilical incision to harvest abdominal fat grafts, which is less noticeable than lower quadrant sites. In patients with large tumors and evidence of a widened sella on preoperative imaging, the use of a lumbar drain may be helpful to prevent CSF leak and promote healing in the immediate postoperative period. Reconstruction of the sellar floor, often with a synthetic or bony graft, is important at the conclusion of the case. In addition to operative approaches targeted at removing the tumor, alternative surgical strategies exist to manage the issues caused by craniopharyngioma cysts and/or resultant hydrocephalus. In most cases, it is our practice to attempt to treat the underlying cause of the hydrocephalus before resorting to a ventriculoperitoneal shunt. In some craniopharyngiomas with large cystic components, placement of a catheter within the cyst can allow the delivery of sclerosing agents, such as bleomycin (an antibiotic that inhibits protein synthesis) or P32 (a radioactive isotope with limited penetration to surrounding tissues). Generally, these catheters are placed with stereotactic guidance because accuracy is of paramount importance. Leakage of toxic agents into the subarachnoid space or normal neural parenchyma is to be avoided. Often, contrast will be instilled through the catheter and an imaging study done to document the absence of a leak before delivery of the sclerosing agent. Following surgical resection of a craniopharyngioma, patients are usually monitored in the intensive care unit. Primary concerns center on the management of hormonal replacement (especially with regard to diabetes insipidus and stress-dose corticosteroids), repeated assessment of visual function (monitoring for hemorrhage or blood pressure–dependent changes in eyesight), and documentation of the level of consciousness (watching for delayed hydrocephalus and effects of hypothalamic injury). As discussed below, postoperative imaging is useful. Continued discussion between intensive care unit staff, nursing, and the neurosurgical team is critical to maximize the likelihood of a successful outcome. Postoperative imaging is critical to assess the extent of tumor resection and to establish a baseline for comparison in future studies. It is our practice to obtain an MRI with and without contrast either intraoperatively (with our operating room scanner) or within 72 hours following surgery. Increasingly, postoperative MR angiography is performed yearly to assess for the development of moyamoya, which is known to occur in this population and responds to surgical treatment if found.38 The presentation of moyamoya can be delayed by years following surgery and should especially be considered in those patients who have also been treated with radiation. MR angiography can also help to identify fusiform dilatation of the carotid arteries, a phenomenon observed in ~10% of children after radical resection that appears to have a benign natural history.39 CT is also performed postoperatively to ascertain whether any calcification remains in the operative bed, a finding considered by some to indicate residual tumor. It is our practice to consider reoperation only if the postoperative mass is unexpected and considered to be safe to resect. Calcified regions of tumor sometimes cannot be safely removed and so may deliberately be left in place. Small flecks of calcium—if found to be the only abnormality present on postoperative imaging—are often observed because their natural history as predictors of tumor recurrence is unclear.40 Endocrine dysfunction is common following the surgical treatment of craniopharyngiomas, with reports of diabetes insipidus occurring in more than 80% of patients and the need for hormone replacement in more than 90% of patients who underwent gross total resections.20,26,27,29,34,35,41,42 Percentages are equally high for the replacement of thyroid hormone, sex hormones (following puberty), and corticosteroids.29 Over half of all surgically treated children are candidates for growth hormone replacement.29 Obesity resulting from hypothalamic injury is present in over half of children following resection.29,36 For many of these patients, the deficits are permanent. As such, long-term endocrinologic follow-up is needed, with appropriate adjustments in hormone replacement (age-specific sex hormones, dosing of growth hormone, and careful monitoring of volume status for desmopressin replacement). Critically important is the recognition of the need for stress-dose corticosteroids in the setting of physiologic challenges such as illness and injury. Increasingly appreciated are the consequences of the surgical treatment of craniopharyngioma on cognitive and emotional function. In the most severe state, akinetic mutism may render patients unable to function in society, but more subtle deficits in intelligence, emotion, and memory may add to the lifetime burden of this disease.30,31 Awareness of these potential problems may facilitate appropriate referral for consultation with neuropsychology, psychiatry, and social work to provide additional support and coping mechanisms for affected patients.20,31 The insidious nature of these deficits may cause them to be overlooked, particularly as the acute issues after surgery settle down with time, and thus mandate continued attention by the family and caregivers. Craniopharyngiomas and the surgery to remove them can both produce long-term deleterious effects on the quality of life of affected patients.30 The wide array of disorders associated with this tumor—endocrinologic, visual, metabolic, cognitive, and oncologic (among others)—has prompted our institution to adopt a model in which patients are treated and followed by a multidisciplinary team comprising specialists able to address each of these issues, a practice shared with other high-volume centers.6,24 We often obtain imaging every 3 to 6 months for the first 2 years, then space out to every 6 to 12 months up to the 5-year post-treatment time point. Several series report that average time to recurrence following operation with gross total resection is 2.5 to 3 years, with ~50% of patients remaining disease-free at 5 years, although these statistics remain controversial, with some reports indicating much better long-term disease-free intervals after gross total resection.1,13,20,36,43 However, craniopharyngioma is notable for its capacity to recur decades after seemingly successful treatment and, as such, warrants prolonged monitoring.24,33 Some reports note a 15 to 40% recurrence of tumor at 15 years, regardless of therapy administered.1,24,44 If recurrence develops, it substantially reduces survival, even in delayed cases (10-year survival without recurrence is =99%, whereas patients with recurrence have a survival of <70% at 10 years).20 We continue to monitor our patients with office visits and MRI studies beyond the 5-year postoperative time point, increasingly spacing out imaging to once every 5 years. If recurrence is detected, the risks and benefits of treatment modalities must be weighed. In younger patients, there is often an impetus to attempt repeat surgical resection to avoid long-term risks of radiation (particularly in preschool-age children). Gross total resection at reoperation has been reported with rates ranging from 30 to 70%.1,33,36,37 Prior radiation therapy and increasing size of tumor are noted as risk factors limiting the success of reoperation.24,33,37 The practice at our institution is to attempt repeat resection if there is documented evidence of growth and the tumor appears to be accessible with minimal morbidity. The wide range of clinical and radiographic presentations of these tumors, coupled with the variety of treatments administered (surgery, radiation, radiosurgery, instillation of sclerosing agents), limits the ability to formulate general guidelines for the management of recurrent disease. As such, many patients are evaluated on a case-by-case basis, emphasizing the importance of providing treatment at an experienced center so that the treating surgeon can more accurately assess the likelihood of success.33,34,37 The management of craniopharyngioma is challenging and best provided by surgeons who routinely treat these tumors at institutions with the resources to administer the required multidisciplinary care. Preoperative assessment should include both MRI and CT, coupled with a complete endocrinologic and visual evaluation. Operative approaches should be selected based on available anatomic routes of access, awareness of preexisting deficits to preserve remaining function, and the surgeon’s experience. The goal of complete resection should be weighed against the risk for severe surgical morbidity and may sometimes prompt the abandonment of attempts at further tumor removal. Follow-up studies include immediate perioperative imaging and endocrinologic replacement as needed. Multidisciplinary care is helpful in the long-term management of these patients. Appreciation of the possibility of delayed recurrence, even decades later, should encourage continued monitoring well after the surgery has finished. The optimal management of patients affected by craniopharyngiomas remains controversial, and it is hoped that ongoing collaborative research will guide clinicians toward increasingly effective treatments.
 Etiology and Pathology
Etiology and Pathology
 Epidemiology
Epidemiology
 Clinical Presentation
Clinical Presentation
 Radiographic Evaluation and Differential Diagnosis
Radiographic Evaluation and Differential Diagnosis
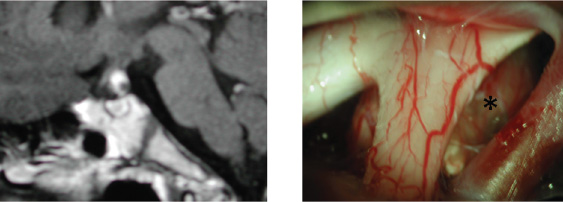
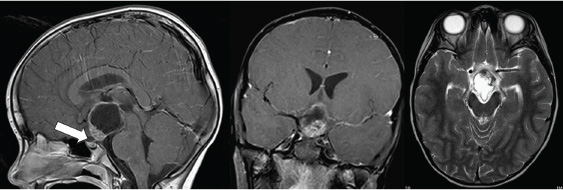
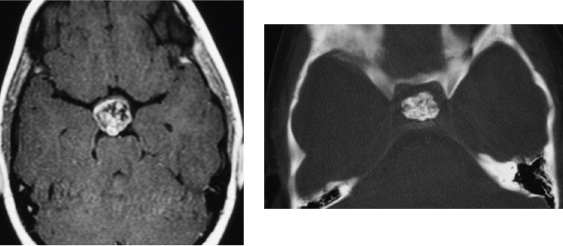
 Initial Evaluation
Initial Evaluation

 Treatment Strategies
Treatment Strategies
 Operative Approaches
Operative Approaches
Preoperative Avoidance of Complications: General Principles
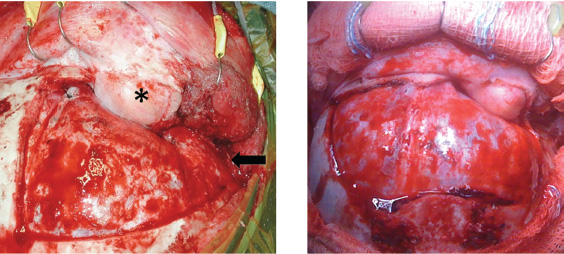
Specific Approaches
Subfrontal
Pterional
Subtemporal
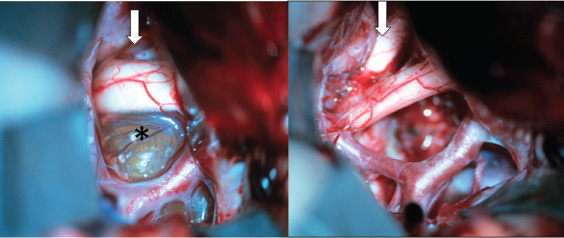
Transcallosal/Transventricular
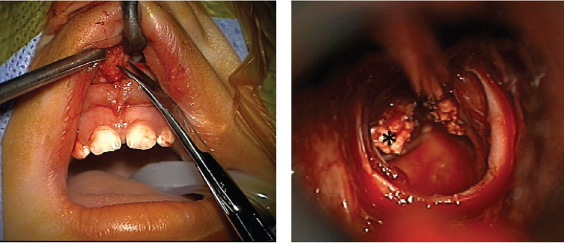
Transsphenoidal (Microscopic and Endoscopic)
Alternative Surgical Treatments
Immediate Postoperative Care
 Outcomes and Follow-up
Outcomes and Follow-up
Imaging
Endocrine, Metabolic, and Cognitive Issues
Long-term Issues and Recurrence
 Conclusion
Conclusion
References

Related posts:
 Introduction and Historical Perspective
Chemotherapy Options for Sellar and Parasellar Tumors
Surgical Treatment of Pituitary Adenomas
Radiologic Evaluation and Diagnosis for Pathology in the Sellar and Parasellar Region
Intracavitary Radiation for Cystic Craniopharyngiomas
Surgical Treatment of Chordomas and Chondrosarcomas
Introduction and Historical Perspective
Chemotherapy Options for Sellar and Parasellar Tumors
Surgical Treatment of Pituitary Adenomas
Radiologic Evaluation and Diagnosis for Pathology in the Sellar and Parasellar Region
Intracavitary Radiation for Cystic Craniopharyngiomas
Surgical Treatment of Chordomas and Chondrosarcomas
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


