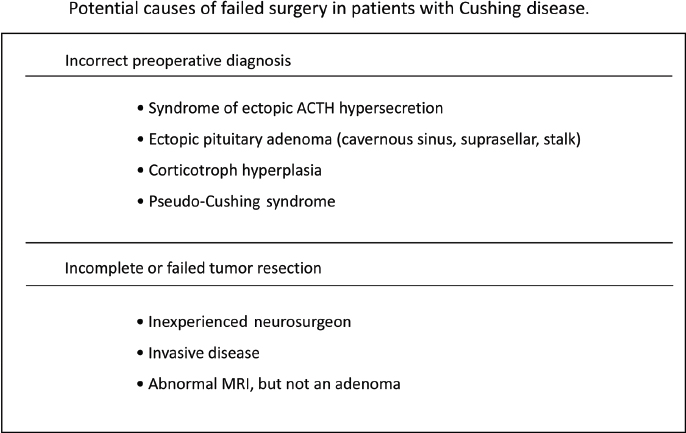
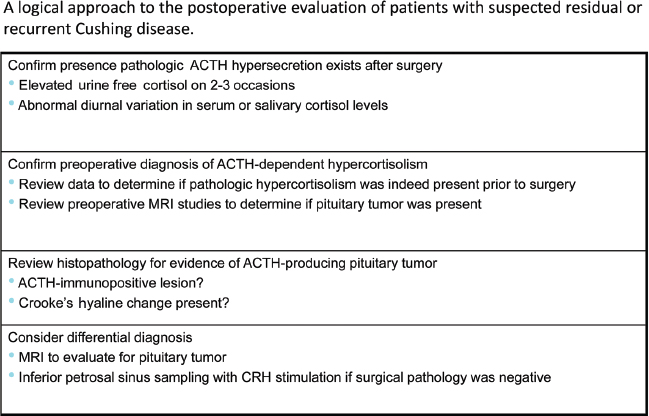
6 Surgical Treatment of Pituitary Adenomas Pituitary adenomas are epithelial tumors arising from the adenohypophysis that can manifest with neurologic symptoms resulting from local mass effects or as a variety of clinical entities depending on the type of hormone secreted. Pituitary adenomas comprise 10 to 15% of intracranial tumors, although the incidence has been noted to be as high as 24% in autopsy series. They are most common in the third and fourth decades of life and overall affect both sexes equally. There are, however, differences in frequency between the sexes for certain subtypes, such as Cushing disease, which is more frequent in women; prolactin-secreting adenomas, which are more common in young women; and null cell adenomas, oncocytomas, and gonadotropin-secreting adenomas, which are more common in men. The classification of pituitary neoplasms has undergone a variety of modifications over time, but they are now commonly stratified based on the hormone they secrete. Endocrinologically, they are considered either active or inactive and are classified as active only if the amount of hormone secreted leads to excessive levels in the blood and clinically evident symptomatology. Inactive adenomas contain the secretory and cellular components necessary for hormone production, but they are not associated with clinical and biochemical evidence of hormone excess. This functional classification is now being further modified to incorporate recent advances in molecular and immunohistochemical staining in pituitary research. The variability of pituitary neoplasms has engendered the need for multiple algorithms of treatment dependent upon the behavior of the classes of tumors themselves. In this chapter, we discuss the indications for surgical treatment as well as the surgical techniques available for the major classifications of pituitary adenomas. Cushing disease is a life-shortening illness caused by hypercortisolism secondary to an adrenocorticotropic hormone (ACTH)–secreting pituitary adenoma. Pituitary surgery is considered the treatment of choice in these cases, providing the greatest likelihood for remission of symptoms, although results thus far have been less than perfect. Recent advances in surgical strategy, endocrinologic assessment, and adjuvant therapies have improved the ability to treat this devastating disease. The large majority of these tumors (89%)1 are microadenomas (<1 cm in diameter) and can be difficult to localize on imaging. In 25 to 45% of cases, magnetic resonance imaging (MRI) does not demonstrate an identifiable neurosurgical target despite biochemical confirmation of pituitary-driven Cushing syndrome.2 In cases in which preoperative imaging is nondiagnostic, preoperative bilateral inferior petrosal sinus sampling (IPSS) with corticotropin-releasing hormone (CRH) stimulation may help to identify the pituitary as the site of pathology. In experienced hands, IPSS preceded by CRH stimulation can correctly lateralize a tumor in 83% of cases in which there is an ACTH gradient of more than 1.4 pg/mL between the two sides.3 In cases in which the lesion is not identifiable on MRI or at the time of microsurgical exploration, the operating surgeon may, depending on the severity of the patient’s symptomatology and expressed desires, opt for hemihypophysectomy in a small percentage of patients or, rarely, complete hypophysectomy with the knowledge that such measures can result in lifelong hypopituitarism requiring hormone replacement and still may not result in remission. Hemihypophysectomy in these cases is usually guided by the results of preoperative IPSS, whereas complete hypophysectomy is usually reserved for patients in whom resolution of the hypercortisolemic state is an absolute necessity. Some surgeons recommend the use of an intraoperative ultrasound device to aid in localization of the adenoma. This author has not found it to be beneficial in our practice.2 We prefer the term remission to cure. In the setting of Cushing disease, remission is defined as resolution of clinical symptoms secondary to hypercortisolemia, biochemical confirmation of normal diurnal cortisol secretion levels, and resumption of normal hypothalamic-pituitary-adrenal function. Initial remission rates following surgery range from 78 to 87% in experienced hands. However, relapse rates of 7 to 11% have been reported following surgery. (Long-term remission rates following surgery have been reported as 42–86%.4) Owing to the relatively high rate of recurrence, much attention in the literature has focused on immediate postoperative factors, which may predict long-term outcomes. In some cases, ACTH and cortisol levels remain unchanged or even may be seen to be elevated in the immediate 24-hour period after surgery. This is most likely attributable to the stress of surgery or anesthesia. However, with a few exceptions in which “late” remission has been documented, levels that remain elevated beyond postoperative day 1 often indicate surgical failure.5 Cortisol levels that decrease to less than 2 to 3 μg/dL within 24 to 72 hours after surgery usually indicate a biochemical remission.6 Simmons et al reported that a comparison of serum cortisol levels measured preoperatively at midnight and ~30 hours after the operation offer the most sensitive indication of long-term remission. With these two measurements, a lower postoperative level offers 95% sensitivity and a 95% positive predictive value for remission up to 2 years following surgery.5 They stress that the administration of exogenous steroids in the immediate postoperative period is unnecessary except in a small percentage of patients with laboratory-documented hypocortisolemia accompanied by symptoms of steroid withdrawal. In fact, routine administration of postoperative steroids often only delays accurate biochemical assessment in the remaining majority of patients. It is also this author’s practice not to routinely prescribe perioperative steroids. Our algorithm requires that patients be closely monitored for signs of addisonian symptoms for 2 days in the hospital, during which time serum cortisol and ACTH levels are sent daily. Patients are subsequently discharged on replacement therapy until the postoperative results become available. If a patient exhibits signs of hypocortisolemia while in the hospital, serum levels are immediately drawn and replacement therapy is initiated without delay. Although they may be dramatically decreased from preoperative values, 1-week postoperative serum cortisol levels within the normal range are not predictive of remission. This finding in fact often signifies trauma to the abnormal ACTH-producing tissue that may eventually lead to divergent outcomes of further necrosis and remission or resumption of secretory activity. In one study of patients with normal serum cortisol levels in the immediate postoperative period, 29% went on to demonstrate long-term remission, whereas 71% demonstrated recurrence from 2 to 4 years following surgery.7 Another indication of viable tumor cells in the immediate postoperative period is a measurable response of serum ACTH level to CRH stimulation. In a series of 82 patients, a normal or exaggerated response predicted recurrence in 18% and 43%, respectively, whereas an absent response can be a predictor of long-term remission at 2-year follow-up.7 Macroadenomas are easier to identify on preoperative imaging and localize intraoperatively. However, they portend a worse prognosis and higher surgical failure rate than microadenomas. In a series of 137 patients with microadenomas at Massachusetts General Hospital, 123 (90%) were cured with transsphenoidal surgery with or without adjuvant therapy. This contrasts with a surgical cure rate in 11 of 17 (65%) patients from the same series who harbored macroadenomas. The 5-year cure rates in this series for patients with microadenomas versus macroadenomas were 96% and 91%, respectively, whereas the 10-year cure rates were 93% and 55%.1 Outcomes have not been found to correlate with the presence of a lesion on preoperative imaging. Remission rates after the initial surgical resection were found to be 79.3% in patients with a normal or inconclusive preoperative MRI, compared with 82.3% of patients with MRI-identifiable adenomas (P = 1.0) in a retrospective review of 248 consecutive patients identified with Cushing disease.8 In many cases, histologically identifiable ACTH-secreting adenomatous tissue is not seen postoperatively. Over the years, there has been much debate as to the significance of this finding. A recent study demonstrated that among patients who underwent transsphenoidal resection for Cushing disease, rates of remission for those with no histologically identifiable tissue were 50%, compared with an 88% remission rate (P <0.001) for those with identifiable pathologic tissue. This is likely due to incomplete extirpation and may necessitate more close observation and follow-up of this particular subset of patients.9 Given the current data, coupled with our own experience, our philosophy has been to rely on the 24-hour urinary free cortisol levels as well as the midnight salivary cortisol levels. If those have been found to be abnormal, confirming Cushing syndrome, then MRI with high-resolution thin cuts through the pituitary sella is performed. If an adenoma is identifiable on MRI, then we proceed to transsphenoidal resection of the tumor. If an ACTH-immunostaining tumor is not found and cortisol and ACTH values remain elevated, an IPSS study with CRH stimulation is done to confirm the diagnosis of Cushing disease. Conversely, if no lesion is seen on the initial MRI, then IPSS studies are done to confirm that the pituitary is the cause of the Cushing disease, as well as to suggest laterality in the gland to help locate the adenoma. We request a 24-hour urinary free cortisol level the day before the IPSS study to ensure that the patient is not eucortisolemic at the time of the IPSS study. The indication for bilateral IPSS (BIPSS) in the preoperative work-up of Cushing disease is controversial. Whereas some centers perform BIPSS in all patients with presumed Cushing disease before transsphenoidal surgery, others do not. We studied this issue, and in our series BIPSS was performed only in patients without a clear adenoma on MRI or if biochemical testing was not fully consistent with Cushing disease. The study, which was a retrospective analysis of 187 adult patients (153 women, 34 men; mean age, 41 ± 13.8 years; range, 18–74 years) with presumed Cushing disease, addressed whether the selective indication for BIPSS in the preoperative work-up of Cushing disease might have affected the early postoperative remission. Early postoperative remission required fasting serum cortisol levels of less than 5 μg/dL and/or urine free cortisol excretion of less than 50 μg/24h. One hundred patients (53.5%) underwent BIPSS before transsphenoidal surgery. Ninety-one of these had either a normal or an inconclusive MRI, and only 9 of the 87 patients with an adenoma on MRI had a BIPSS. Of all patients, 175 (93.6%) showed an early postoperative remission. Of the 100 patients with a preoperative BIPSS, 94 (94%), and of the 87 patients without a BIPSS, 81 (93.1%) had a postoperative remission. All patients without BIPSS and no early remission had an ACTH-positive adenoma on pathology, and therefore we suggested that the performance of a BIPSS would not have changed the management of these patients. In summary, 93.6% of all patients had an early postoperative remission. The selective indication for BIPSS in the preoperative work-up of Cushing disease did not negatively affect the outcome and thus should be considered more broadly.8 Patients who demonstrate elevated postoperative ACTH and cortisol levels are candidates for attempted surgical re-exploration. In addition, some authors advocate repeat surgery in the immediate postoperative period when cortisol levels are within the normal range, given the high rate of recurrence. They argue that in early re-exploration, the lack of scar formation and the surgeon’s acquaintance with the surgical target facilitate access and resection. In all cases of repeat surgery, the biochemical and radiologic work-up must be rescrutinized and possibly repeated to verify a pituitary origin of disease. Mandatory testing in this setting includes IPSS for ACTH with CRH stimulation if not previously performed. A small percentage of patients may in fact harbor an ectopic source of abnormal ACTH-secreting cells, such as within the cavernous sinus, that would not be amenable to repeat surgery.10 Notwithstanding such cases, if no discrete adenoma is found at the time of repeat exploration, the surgeon may opt for hemihypophysectomy or complete hypophysectomy, as previously discussed. Remission rates following repeat surgery for residual or recurrent disease range from 30 to 53%.1,11 Hypopituitarism, as well as complications including infection and cerebrospinal fluid (CSF) leaks, are more common following repeat surgery, most likely because of the tendency to perform more aggressive procedures. This author uses a wide transnasal transsphenoidal exposure, opening from carotid canal to carotid canal (cavernous sinus to cavernous sinus) with either a microscope or endoscope technique. If the lesion was visible on the MRI, then we proceed to the area of the adenoma. If there was no visible lesion on the MRI, then we start on the side with the higher ACTH values on IPSS. Exploration begins between the gland and the dura. If no tumor is visible between the gland and dura, we then incise into the gland vertically on the suspected lateral wing of the gland (Fig. 6.1). If tumor is found, it is removed gently with the ring curettes and microdissectors. At this point, if the adenoma is still not visible, a vertical incision is made into the opposite lateral wing vertically. If adenoma is not identified, then a horizontal incision is made across the lower third of the anterior surface of the gland and the depth of the gland is explored. If no tumor has been seen and the IPSS study shows strong laterality, a hemihypophysectomy is done on that side. If remission is not achieved after the first exploration and testing is definitive for a pituitary etiology, a second exploration is performed more vigorously, with the understanding that hypopituitarism may develop. If two surgical procedures fail to yield remission, adjunctive therapy such as stereotactic radiosurgery and/or medical therapy with ketoconazole is recommended (Figs. 6.2 and 6.3). Acromegaly is a disease of chronic overproduction of growth hormone (GH). In the child and preadolescent, excess GH leads to abnormally increased height and gigantism as well as organomegaly. Once the epiphyseal plates have closed, the manifestations are those of acromegaly. The consequences of GH hypersecretion are numerous. Bone and soft-tissue overgrowth is almost always present, resulting in changes in shoe size and ring size and a coarsening of the facial features, with enlargement of the lips, macroglossia, a prominent brow, prognathism, and increased spaces between the teeth. A low voice is a product of laryngeal hypertrophy. Patients often go on to develop skeletal abnormalities secondary to new bone growth, including vertebral body osteophyte formation leading to spinal stenosis and bowing of the long bones. Overgrowth contributes to a significant increase in weight and stress on the joints, cartilaginous erosion, and severe osteoarthritis. More seriously, acromegaly causes numerous cardiovascular complications, such as cardiomyopathy, arrhythmias, and coronary artery disease. GH elevation contributes to markedly increased rates of diabetes and hypertension. Mortality is significantly increased in patients with untreated or uncured acromegaly. The goals of treatment are control of GH and insulin-like growth factor 1 (IGF-1) levels, such that GH levels are less than 1 μg/L after an oral glucose load and IGF-1 levels are normal, adjusted for age and sex. The tumor mass needs to be removed or significantly reduced, such that the optic nerves are free of compression or impending threat of such.12 It is common for patients with acromegaly to require multimodality therapy.13 Fig. 6.2 Potential causes of failed surgery in patients with Cushing disease. Transsphenoidal microsurgery is the preferred primary treatment for acromegaly, although rarely craniotomy may be necessary in the case of certain tumors that have a significant extrasellar or suprasellar component.14 Surgical resection of the adenoma provides the greatest hope for cure and is generally safe, with acceptable morbidity.15 As one would expect, tumor size has clearly been shown to influence the likelihood of remission. The success rate for modern microadenoma resection alone ranges from 72 to 88% in this author’s series. Success rates for operating on macroadenomas are expectedly lower than those for smaller tumors. Macrodenomas that are noninvasive have modestly lower remission rates of 50 to 56%.14 Those tumors found to be invading the cavernous sinus or surrounding structures have a further reduced rate of remission. Major decreases in remission exist when the tumor is invasive and larger than 20 mm. GH levels may be predictive of the likelihood of success. According to Shimon et al, whereas patients with preoperative GH levels of less than 20 μg/L and 20 to 50 μg/L had cure rates of 90 and 79%, respectively, those with GH levels of more than 50 μg/L were cured at a rate of 16%.16 Similar data by Freda et al found 90 and 80% cure rates for microadenomas and macroadenomas, respectively, when the preoperative GH level was less than 10 μg/L; these fell to 55% for patients with GH levels of of 30 to 50 μg/L. No patients were cured who had a preoperative GH level of more than 200 μg/L.17 Because GH levels rapidly fall following resection, while IGF-1 can take a longer time to normalize, GH levels are routinely the test of choice in the immediate postoperative period. Early GH levels can be predictive of success or failure. There is not a clear cutoffin the early postoperative laboratory results to define cure, but GH levels of less than 2 μg/L on the first postoperative day have been found to correlate with surgical success in 99% of patients.18 Accordingly, only 1 of 65 patients with a GH level of more than 2 μg/L on postoperative day 1 was later determined to have biochemical cure.18 Dural invasion and extrasellar extension of tumors have been shown to be associated with persistent disease. Patients with acromegaly usually present because of their endocrinologic disturbances. Some patients, however, present with visual disturbances. The majority of patients recover some degree of visual function following transsphenoidal surgery. The recovery of visual function relates to the duration of optic nerve compression and the ability to adequately decompress the nerves in the operating room. As patients with acromegaly largely have other symptoms as well, they are more likely to be diagnosed at an earlier point in their disease. Nonetheless, some patients do present with significant nerve compression and/or pituitary apoplexy leading to acute visual deterioration. In one study of patients with acromegaly and optic nerve compression at the time of surgery, 75% had an improved visual examination following transsphenoidal decompression.15 There is a small subset of patients who will not present until their elderly years. As many as 3 to 5% of patients with acromegaly do not present until they are older than 65 years of age.19 Many clinicians are tempted to steer these patients away from surgical resection because of the belief that they are unable to handle the stresses of surgery. Minniti et al, in a series of 22 patients with acromegaly past the age of 65, reported no significant perioperative morbidity and no mortality due to transsphenoidal resection. Sixty-eight percent of patients were reported cured based on biochemical criteria and significant improvement in cardiovascular function and glucose tolerance.19 Transsphenoidal surgery for GH-secreting tumors is very safe. In general, the morbidity associated with surgical resection of tumors in patients with acromegaly mirrors that of surgery in patients with other tumors via the same method. Anesthetic considerations may be somewhat different if there is significant airway compromise. The soft-tissue, cartilaginous, and bony overgrowth associated with the disease may contribute to a narrowed surgical corridor, especially when a transnasal route is employed, making instrument manipulation even more challenging. Another consideration for surgical planning in these patients is the significant narrowing of the inner borders of the carotid sinus giving rise to a reduced intercarotid distance, which, if neglected, may result in an increase in vascular injury.20 In experienced hands, major complications such as visual impairment, severe meningitis, and death have been reported in fewer than 2% of cases. CSF leaks, hypopituitarism, and diabetes insipidus have been reported in roughly 5% of postoperative patients. The risks of transsphenoidal surgery as well as the likely rates of cure reported in this chapter are predicated on the procedure being done by an experienced surgeon.21–24 Reoperation for acromegaly is a controversial subject. Rates of surgical success in these patients have been historically quite poor. Recent experience by dedicated pituitary surgeons has been quoted as 44 to 76%.25 This can most likely be attributed to extensive tumor involvement, given that at the time of surgery 70 to 80% of tumors are macroadenomas, which are less amenable to complete surgical resection.26 The postoperative patient with persistent disease is a complex clinical entity. There is much controversy surrounding the ideal course of treatment after surgical failure. Goals of therapy are fourfold: to gain biochemical control, reverse medical comorbidities and risk for mortality, relieve signs and symptoms of the disease, and limit local tumor mass effects.27 Some patients need reoperation for progressive visual deterioration. In general, surgical re-exploration is reserved for those who on follow-up demonstrate persistent compression of the optic pathway or in whom residual tumor may have become more surgically accessible over time. It has been noted that patients undergoing reoperation for impairment do not generally seem to garner as much visual improvement as those undergoing a primary operation with the same complaint. What defines recurrence of active acromegaly is a confusing issue. As testing becomes more sensitive, more patients are noted to have subtle signs of endocrinologic laboratory abnormality. Some patients now considered to have recurrence may just have had mild GH dysfunction that was not detected by earlier, less accurate tests. We have previously reported on a subset of patients with postoperative IGF-1 levels that normalized but failed to be suppressed to less than 1 μg/L. Five of these 14 patients later developed evidence of elevated IGF-1.28 This suggests a group of patients with subtle endocrinologic dysfunction who, although they have normal IGF-1 levels and hence are technically in remission, probably still have some latent disease. Although no intervention is considered necessary provided the IGF-1 remains under control, this group may benefit from closer monitoring. The current treatment paradigm for acromegaly involves a multidisciplinary approach (Fig. 6.4). It is incumbent on the patient’s primary care doctor to notice the signs and symptoms of the disease early and refer the patient for definitive treatment. Success correlates with pretreatment GH levels. To reduce the patient’s risk for mortality to baseline levels, the IGF-1 level must be normalized and the GH level suppressed to less than 1 μg/L following an oral glucose tolerance test. Surgery offers the best possible opportunity for cure. In experienced hands, cure rates for patients with a microadenoma approach 90%; those with larger and/or invasive tumors do not fare quite so well. Patients who do not have full normalization of their IGF-1 level postoperatively should be considered for further therapy or reoperation. Reoperation can be considered, but rates of cure are significantly lower than in the primary surgical group. Medical therapy is successful in lowering GH and IGF-1 levels and may be employed both presurgically and postsurgically, but its effects are sustained only during the course of administration. New, long-acting depot forms of somatostatin analogues are available that offer equal efficacy and longer dosing intervals. The newer GH antagonist holds further promise in the medical armamentarium, but patients’ GH levels can no longer be followed. Radiation is a good treatment option for patients with uncured acromegaly following surgery. The cure is slow to develop, but more than 50% of patients may achieve normalized IGF-1 levels this way. Both radiation therapy and stereotactic radiosurgery carry significant risks of panhypopituitarism necessitating hormone therapy. Similar to the prolonged time to cure, patients have a gradual onset of pituitary hormone dysfunction, should it develop. The incidence of difficult intubation in patients with acromegaly has been reported to vary from 10 to 30%.29–31 Airway difficulty has been attributed to morphologic changes produced by excess GH. These changes include prognathism, macroglossia, and thickening of the pharyngeal and laryngeal soft tissue. Thickening of the vocal cords, recurrent laryngeal nerve paralysis, narrowing of the cricoid arch, and hypertrophy of aryepiglottic and ventricular folds have also been cited as causes of difficult intubation in patients with acromegaly.32 Of our patients with acromegaly and a preoperative evaluation of a difficult airway, 61% proved to be difficult intraoperatively. Conversely, 91% of patients with GH-secreting tumors who required three or more attempts at intubation were felt to have a “difficult” airway preoperatively. Our 17.3% rate of difficult intubation is well within the range of difficult airway in acromegaly of 10 to 30% reported in the literature. Patients with acromegaly were 4.4 times more difficult to intubate than were patients with non–GH-secreting/non–ACTH-secreting tumors. Although considered the gold standard, fiberscopic intubation was only 85% successful in securing the airway (Fig. 6.5). Surgical techniques, whether via a microscopic or endoscopic venue, are the same as for other pituitary adenomas. There is no need to explore the gland, as is necessary with Cushing disease, because no patient whom we operate on has a normal MRI. Because of the potentially narrowed distance between the carotid arteries, a micro Doppler probe is often used before the dura is incised. Prolactinomas are the most commonly diagnosed pituitary adenomas, representing ~30% of cases.33 Rarely life-threatening, they cause symptoms primarily as a result of hyperprolactinemia, which in turn results in alterations in reproductive and sexual function and may also cause symptoms related to mass effect. This is the pituitary tumor for which a reliably effective medical treatment is most often available and that in most cases is the favored primary therapy. Surgical treatment, however, has proved to provide similar results as in other endocrine-secreting adenomas and is applicable as the appropriate intervention for prolactinomas in certain cases. Lactotropes are controlled by a negative dopaminergic effect from the hypothalamus, and based on this, dopamine agonists have become the standard medical therapy. Dopamine agonists have been remarkably effective in both reducing the size of the tumor and normalizing serum prolactin levels below the accepted remission level of 20 ng/mL. Bromocriptine was the first of these medications, but more recently others, such as cabergoline, quinagolide, lisuride, pergolide mesylate, and terguride, are also used. Dopamine agonists have been shown to have a high response rate, with reports of normalization of prolactin in 70 to 80%, tumor shrinkage in 80 to 90%, and restoration of ovulation in 80 to 90%.34 Based on these results, some would argue that medical treatment should be used in all patients except the few with significant side effects from medical treatment. In addition, in an important study, it has been shown that a majority of patients who respond to cabergoline with normalization of prolactin levels and a reduction in tumor size may experience remission of hyperprolactinemia following discontinuation of the drug.33 This study suggests the potential for a curative treatment with medical therapy in a selected group of patients. Dopamine agonists do have some significant disadvantages that may temper their use in all patients. The effects on the tumor are reversible, making the therapy lifelong in most cases and thus requiring significant compliance. Additionally, there can be significant side effects, including nausea, vomiting, postural hypotension, headaches, depression, and anxiety, that can make long-term compliance even more difficult in some patients. In cases of pituitary apoplexy and in tumors with large cystic components, dopamine agonists are not effective in effecting shrinkage. A group of bromocriptine-resistant prolactinomas with increased aggressiveness and increased incidence in male patients has been recently described.35 There have been recent reports of an association between valvular heart disease and dopamine agonists in patients being treated for Parkinson disease. However, these patients with Parkinson disease were receiving doses approximately seven times higher than the maximal dose used for the treatment of prolactinomas.12 In a review of the current literature on the subject, Valassi et al reported that most published articles on the subject do not show an association between the use of dopamine agonists and valvulopathy; however, they recommend using the lowest dose possible and that the physician address the use of echocardiographic monitoring on a case-by-case basis.36 The role of surgery in the treatment of prolactinomas has been under discussion. Some argue that surgery should be the initial treatment of microprolactinoma, with medical therapy as an adjunct only in cases without remission.37 The rate of long-term remission varies significantly, however, depending on the size of the tumor and preoperative prolactin levels. The most favorable results have been demonstrated in patients with microadenomas and levels below 200 ng/mL, with remission rates ranging from 50 to 84% in long-term follow-up.34,37,38 The remission rate drops with macroadenomas and with prolactin levels from 200 to 500 ng/mL, even though these tumors are amenable to surgical resection. Surgery has not been shown to be useful alone in patients with giant adenomas or tumors with prolactin levels higher than 500 ng/mL, in whom the remission rate decreases to zero. These lesions are better treated with medical therapy, possibly in conjunction with surgery and radiation. Overall, a lower prolactin level portends a greater chance of long-term remission. Based on the most current literature, surgical removal can be recommended for most patients with a macroadenoma and a prolactin level of less than 200 ng/mL, with anticipation of a high remission rate. Patients with tumors larger than 2 cm and/or a prolactin level below 500 ng/mL should first treated with a dopamine agonist to reduce the tumor volume and then undergo surgical resection. Any residual tumor can then be treated medically. For patients with very large or invasive tumors or tumors with a prolactin level above 500 ng/mL, medical therapy should be the primary treatment modality. Another consideration in the management of these patients is the incidence of spontaneous CSF leak after the medical treatment of macroprolactinomas, which is a well-known phenomenon. A recent study of a small series of patients found that those subjects with adenomas invading the sphenoid sinus were at the highest risk for spontaneous CSF leaks after medical treatment that required neurosurgical repair.39 The subgroup requiring specific discussion comprises patients who are pregnant and harbor prolactinomas. If the patient becomes symptomatic, a dopamine agonist can be administered. The data regarding the effects of continuous dopamine agonist therapy on the fetus are limited but suggest no ill effect. During pregnancy, surgery should be undertaken only if the tumor does not respond to medical treatment and if there are progressive neurologic symptoms. Patients with macroadenomas who desire pregnancy should undergo a transsphenoidal resection before conception or remain on bromocriptine during the pregnancy. Adenomas secreting thyroid-stimulating hormone (TSH) are the least common of the pituitary tumors, representing only 1 to 2% of cases. Before any surgical intervention, the hyperthyroidism should be controlled to minimize the risk for cardiac arrhythmias that may occur during induction of anesthesia or intraoperatively. This is commonly done with a preoperative β blocker. If the surgery is nonemergent, an antithyroid drug (propylthiouracil or methimazole) can be added. Transsphenoidal surgery is the primary treatment but is associated with cure rates of only ~35 to 62% when performed alone and 55 to 81% when performed in conjunction with medical treatment and radiotherapy.40–42 The criteria for cure in TSH-secreting adenomas have not yet been well defined. Radiosurgery or radiation therapy can be used if clinical remission is not obtained with surgery alone. However, radiosurgery and radiotherapy are not typically used as a primary treatment modality. Approximately one-third of pituitary tumors are considered nonsecreting (also called nonfunctioning) adenomas. They are generally associated with older age, with a peak in the fifth decade. Patients usually present with signs and symptoms of hypopituitarism, and commonly with associated visual field defects and other ophthalmologic problems. Despite their similar clinical presentation, endocrine-inactive tumors are a heterogeneous group. Ultrastructural studies have demonstrated that these tumors actually contain secretory granules and that a majority of them synthesize follicle-stimulating hormone (FSH), luteinizing hormone (LH), and the α subunit, with a smaller subset producing other anterior pituitary hormones that do not have clinical manifestations.43 No clear and consistent difference in treatment or prognosis has emerged among the various subtypes, however. Because endocrine-inactive tumors do not cause hypersecretory syndromes, patients usually present with symptoms associated with mass effect. Patients usually describe headaches, visual changes, and symptoms of pituitary insufficiency. In a majority of cases, the tumor is a macroadenoma at the time of diagnosis. Surgery remains the primary treatment of patients with inactive adenomas. The goals are to relieve the mass effect, restore pituitary function, and obtain a tissue diagnosis. As with other pituitary adenomas, the preferred approach is transsphenoidal, but cases with significant eccentric extension outside the sella may require a transcranial approach. The results of surgery for endocrine-inactive tumors are not as clearly described as those of surgery for other adenomas because there are no clear criteria for cure. Studies have consistently demonstrated that 70 to 80% of patients experience significant improvement in visual function. In addition, nearly 100% of patients report resolution of headaches, and there is an improvement in pituitary function ranging from 15 to 57%, depending on the series and the criteria for hypopituitarism.44 Clinical improvement is not suggestive of complete tumor removal. Postoperative imaging a few months after surgery is required to assess the extent of tumor resection. Early imaging may not be as helpful or accurate because of confounding edema, postoperative hemorrhage, and hemostatic material. This study then acts as a baseline for future comparison. We do not discuss adjunctive therapy for residual tumor in this chapter. Rather, it is discussed in other chapters in this book. The historical development of transsphenoidal surgery is a complex tale of innovative leaps in ideology coupled with periods of extensive surgical experimentation and even complete rejection of the technique. Summaries of the historical movements that have resulted in its adoption as the preferred approach to tumors of the hypophysis have already been well documented. However, within this history there exist some significant revolutions in technique and technology that have further advanced the utility of this operation and merit further study. The first of these revolutions was in surgical technique, with the introduction of sublabial and transnasal approaches to the pituitary. Then, with the introduction of intraoperative image intensification by Gerard Guiot in 1958 and the surgical microscope by Jules Hardy in 1967, many of the technical difficulties faced by pioneers such as Harvey Cushing could be circumvented. This laid the foundation for the advances that now shape modern transsphenoidal surgery, including the use of endoscopy, intraoperative imaging, frameless guidance, and radioimmunoassay. Currently, the transsphenoidal approach is the preferred approach for more than 90% of pituitary tumors. Occasionally, a transcranial approach becomes necessary for those very large or asymmetric tumors with minimal sellar content. The transsphenoidal procedure is reasonably similar whether a microscopic or endoscopic technique is used. The microscope gives a clear three-dimensional view, whereas the endoscope gives a far more extended view (Figs. 6.6, 6.7, 6.8, and 6.9). With a microscope, the average exposure of the sella is ~12 mm in width and from the tuberculum to the clivus in the vertical plain. Generally, the carotid canals can be visualized. In the sphenoid sinus, the endoscope allows visualization of the entire inner surface of the sphenoid sinus, which is far more important in the extended transsphenoidal operation for tumors other than adenomas, such as meningiomas and craniopharyngiomas. Within the sella, the endoscope allows more lateral inspection of the cavernous sinus walls as well as the suprasellar region, provided that the arachnoid membrane and diaphragma sellae have not completely descended into the sella with tumor decompression. We have used both approaches and often add the endoscope as an adjunct when we use the microscope. During an endoscopic procedure, a speculum may be inserted so that the microscope can be added as an adjunct. Clearly, both techniques are excellent, and the surgeon may have both available and should be adept with both. Fig. 6.6 An endoscopic view through the right naris of the ostium into the sphenoid sinus. For anatomic localization and direction to the sella, fluoroscopy was the mainstay but offered only one plane, lateral, and entailed the problem of radiation exposure. Newer techniques with MRI- or CT-directed BrainLab/ Stealth equipment may make fluoroscopy unnecessary. These techniques offer the advantage of multiplane views as well as lower or no radiation exposure. There are numerous studies showing that in practiced hands, these techniques do not add substantially to the operative time and may reduce complications, such as carotid artery injury, of migrating from the midline approach. A small error in the angle toward the sella during exposure can lead directly to the carotid canals. Fig. 6.7 Endoscopic view of the sellar floor and lateral carotid canal. We operate with the patient in the supine position and the head just slightly flexed and tilted toward the left because the surgeon is generally on the patient’s right side. With an endoscopic approach, the head may be kept straighter because a four-handed technique with one surgeon on either side of the patient may be advantageous. The right lower corner of the abdomen is also prepared in case there is need of a fat graft to seal an exposure of the CSF space. Either the fluoroscope or BrainLab equipment is positioned for localization and direction. Fig. 6.8 Endoscopic view of the pituitary. Fig. 6.9 Transsphenoidal endoscopic surgery. A transnasal submucosal exposure of the front of the sphenoid sinus is created with use of the vomer to ensure the midline if fluoroscopy is used. The orifices into the sphenoid are enlarged, allowing exposure of the back wall of the sphenoid sinus and anterior sella wall. If the sella is dramatically enlarged, or if it is anticipated that a large opening into the CSF space will be created, such as with an extended procedure, a pedicled mucosal flap along the midline is created and tucked aside in the nasal cavity for closure at the end of the procedure. We lean more toward a microscopic approach and therefore insert a bivalved microspeculum through the nares with one blade on either side of the vomer beneath the mucosal flaps. In the past, when a sublabial exposure was done with a larger speculum, care was taken to avoid putting the blade of the speculum into the sphenoid sinus for fear of fracturing the medial walls of the orbits. With a transnasal approach and a microspeculum, we tend to place the speculum just inside the sphenoid sinus wall and open it gently to avoid fractures. This gives better exposure and keeps the speculum from rotating out of the vertical position. With an endoscopic technique, a speculum may be unnecessary. Once the anterior sellar wall is exposed, it may be opened with a microdissector if it is thin or with a drill or osteotome if it is thick. It must be opened to the medial wall of cavernous sinus on both sides. This can be confirmed with BrainLab or visually by a change in direction of the dura as well as a color change. A micro Doppler probe may be helpful in confirming that the exposure is not too far lateral, exposing the carotid artery to injury. The dura can be opened sharply in an X fashion or with excision of a large rectangle anteriorly. If tumor is clearly invading through the dura, the latter opening is used to excise the invaded dura. A vertical cross-opening yields less exposure of the gland. Tumor dissection varies with the size of the adenoma. With macroadenomas, the tumor is usually evident upon opening the dura. With microadenomas, dissection is guided by the preoperative imaging studies. With both microadenomas and macroadenomas, a clear plane can be found between the adenoma and the normal gland. This is the plane we follow to completely excise the tumor. We advise removing the tumor that is central, inferior, and lateral before approaching the suprasellar portion. If the superior tumor is removed first, the diaphragma sellae and arachnoid may herniate into the sella, significantly obscuring the field. At this point, the endoscope may be of great value to inspect the medial walls of the cavernous sinuses as well as the suprasellar walls to be as certain as possible that no further tumor exists. A wider view with an angled endoscope is an advantage. After the adenoma has been resected, closure is somewhat dependent upon whether CSF has been seen. We ask anesthesia to perform a Valsalva maneuver to raise the intracranial pressure to see if CSF leakage is present. If CSF is not seen at any time during the procedure, we do not routinely use fat or other substances to fill the sella. If the sella has been markedly eroded, with a large expanse of arachnoid seen in the sella, then fat may be placed in the sella regardless of whether CSF was seen. The sella floor is closed with a piece of vomer bone that was removed during the exposure. If an adequate piece of bone is not available, then a biodegradeable plate is used instead. The bone or plate is wedged under the edges of the opened sellar wall. If the erosion of the sellar wall has been so extensive that no edges exist, this cannot be done. If there has been CSF leakage during surgery, the back of the sphenoid sinus is packed with fat harvested from the abdomen. We rarely place a spinal drain for CSF diversion either before or after surgery. Once the speculum is removed, the mucosa is reapproximated to the midline and a Mercilene sponge is placed in the nares for 48 hours. Patients are hospitalized for 2 days on our service and are generally back at full activity within 1 week. Pituitary adenomas are relatively common tumors within the sellar and parasellar region. Surgical resection represents an important part of the treatment paradigm for patients with hypersecretory adenomas and nonsecretory adenomas exerting mass effect. Radiologic and endocrine results after pituitary adenoma surgery are favorable. Long-term follow up of these patients is required to detect recurrence. Reoperation generally affords less favorable results than the initial surgery.
 Surgical Management of Cushing Disease
Surgical Management of Cushing Disease
Repeat Surgery
Surgical Technique
 Surgical Management of Acromegaly
Surgical Management of Acromegaly
Surgical Technique
 Surgical Management of Prolactinomas
Surgical Management of Prolactinomas
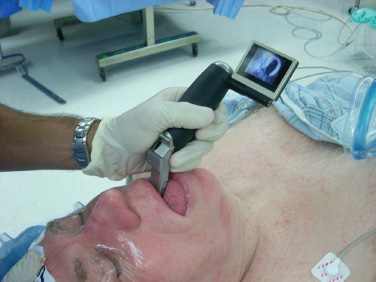
 Surgical Management of Adenomas Secreting Thyroid-Stimulating Hormone
Surgical Management of Adenomas Secreting Thyroid-Stimulating Hormone
 Surgical Management of Nonsecreting Pituitary Adenomas
Surgical Management of Nonsecreting Pituitary Adenomas
 Surgical Technique for Pituitary Adenomas
Surgical Technique for Pituitary Adenomas
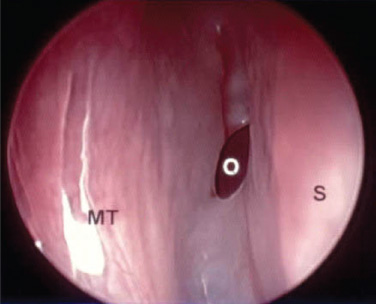
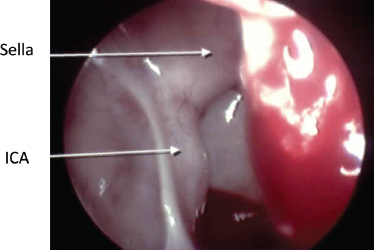
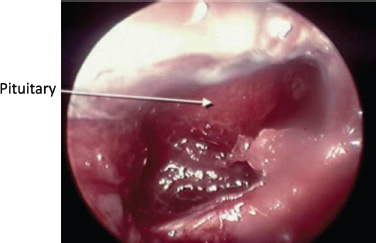

 Conclusion
Conclusion
References
Related posts:
 Introduction and Historical Perspective
Chemotherapy Options for Sellar and Parasellar Tumors
Stereotactic Radiosurgery
Radiologic Evaluation and Diagnosis for Pathology in the Sellar and Parasellar Region
Intracavitary Radiation for Cystic Craniopharyngiomas
Surgical Treatment of Chordomas and Chondrosarcomas
Introduction and Historical Perspective
Chemotherapy Options for Sellar and Parasellar Tumors
Stereotactic Radiosurgery
Radiologic Evaluation and Diagnosis for Pathology in the Sellar and Parasellar Region
Intracavitary Radiation for Cystic Craniopharyngiomas
Surgical Treatment of Chordomas and Chondrosarcomas
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


